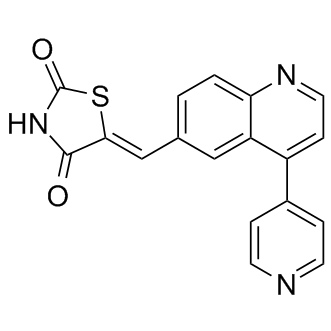
Mekinist (trametinib)
JTP-74057, GSK212, GSK1120212
N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , 4 , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4, 3-d] pyrimidin-1-yl] phenyl}acetamide
| Molecular Weight | 615.39 |
| Formula | C26H23FIN5O4 |
| CAS Number | 871700-17-3 |

Trametinib (GSK1120212) is experimental cancer drug. It is a MEK inhibitor drug with anti-cancer activity.[1]
It inhibits MEK1 and MEK2.[1]
Trametinib had good results for V600E mutated metastatic melanoma in a phase III clinical trial.[2]
- Trametinib, NCI Drug Dictionary
- METRIC phase III study: Efficacy of trametinib (T), a potent and selective MEK inhibitor (MEKi), in progression-free survival (PFS) and overall survival (OS), compared with chemotherapy (C) in patients (pts) with BRAFV600E/K mutant advanced or metastatic melanoma (MM).
May 29, 2013 —
GlaxoSmithKline plc announced today that the U.S. Food and Drug Administration (FDA) has approved Mekinist (trametinib) as a single-agent oral treatment for unresectable or metastatic melanoma in adult patients with BRAF V600E or V600K mutations. Mekinist is not indicated for the treatment of patients who have received a prior BRAF inhibitor therapy. The mutation must be detected by an FDA-approved test, such as the companion diagnostic assay from bioMérieux S.A., THxID™-BRAF.
Among those with metastatic melanoma, approximately half have a
BRAF mutation, which is an abnormal change in a gene that can enable
some melanoma tumours to grow and spread.
Mekinist is approved for patients with the BRAF V600E mutation, which
accounts for approximately 85 percent of all BRAF V600 mutations in
metastatic melanoma. It is also approved for patients with the V600K
mutation, which makes up approximately 10 percent of all BRAF V600
mutations in metastatic melanoma.Melanoma is the most serious and deadly form of skin cancer.[iii] According to statistics from the National Cancer Institute, in 2013 there will be an estimated 9,480 deaths resulting from melanoma in the United States.[iv] When melanoma spreads in the body, the disease is called metastatic melanoma.[v] Approximately half of all people with metastatic melanoma have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread.2 One in two patients worldwide with metastatic melanoma is expected to survive for a year after diagnosis,while in the U.S., the five-year survival rate was 16 percent (2003-2009). The median age of a newly diagnosed metastatic melanoma patient is almost a decade younger than other cancers.
Mekinist (trametinib) is now approved for the treatment of adult patients with unresectable or metastatic melanoma with BRAF V600E and V600K mutations as detected by an FDA-approved test. Limitation of use: Mekinist is not indicated for the treatment of patients who have received a prior BRAF inhibitor therapy.
(WO2005121142A1). Aniline a reaction with CDI was added cyclopropylamine get two , two and malonic acid cyclization get 3 . 3 chlorination with phosphorus oxychloride reaction with methylamine 4 , as well as byproducts 5 (ratio of 2:1). Mixture 4 + 5 and acid 6 crystals obtained after cyclization compound 7 (pure substance). 7 with activated trifluoromethanesulfonyl chloride to the amide 8 SNAr reaction occurs 9 , 9 in alkaline conditions rearrangement trimetazidine imatinib.


…………………
http://www.google.com/patents/WO2014039375A1?cl=en
The term “trametinib” as used herein means the MEK inhibitor represented by the structure of formula (I):

Depending on naming convention, the compound of formula (I) may also properly be referred to as N-{3-[3-cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-6,8-dimethyl-2,4,7- trioxo-3,4,6,7-tetrahydropyrido[4,3-d]pyrimidin-1(2H)-yl]phenyl}acetamide.
Trametinib is disclosed and claimed, along with pharmaceutically acceptable salts thereof, and also as solvates thereof, as being useful as an inhibitor of MEK activity, particularly in treatment of cancer, in WO 2005/121 142. Trametinib can be prepared as described in WO 2005/121 142.
Suitably, trametinib is in the form of a dimethyl sulfoxide solvate. Suitably, trametinib is in the form of a sodium salt. Suitably, trametinib is in the form of a solvate selected from: hydrate, acetic acid, ethanol, nitromethane, chlorobenzene, 1 -pentancol, isopropyl alcohol, ethylene glycol and 3-methyl-1 -butanol. These solvates and salt forms can be prepared by one of skill in the art from the description in WO2005/121 142.
…………………….
http://www.google.com/patents/WO2005121142A1?cl=en
Example 3-10 By treating N-{3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , , 7-trioxo-3 ,4,6, 7-tetrahydro-2H- pyrido [4 , 3-d]pyrimidin-1-yl] phenyl Jmethanesulfonamide 46 according to conventional methods, sodium salt and potassium salt thereof were obtained.
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1- yl]phenylJmethanesulfonamide sodium salt:
^-NMR (DMSO-de, 300 MHz) δ 0.47 (brs, 2H) , 0.70-0.90 (m, 2H) , 1.23(s, 3H) , 2.35(brs, IH) , 2.82(s, 3H) , 3.22(s, 3H) , 6.69(t, J=8.8Hz, IH) , 6.81 (d, J=8.1Hz , IH) , 6.98 (s, IH) , 7.02 (d, J=8.8Hz , IH) , 7.10-7.30 (m, 2H) , 7.38(d, J=9.2Hz , IH) , 10.22(brs, IH) . MS (ESI) m/z 652 [MH]+.
N-{3-[3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1- yl]phenylJmethanesulfonamide potassium salt: Example 4-1
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 ,8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d]pyrimidin-1-yl] – phenyl}-acetamide Step 1 Synthesis of l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) rea


Step 3 Synthesis of 6-chloro-3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -lH-pyrimidine-2 , -dione

2, 4, 6-trione 49 (59.0 g) obtained in Step 2 were added phosphorus oxychloride (85.0 ml) and dimethylaniline (29.0 ml), and water (8.3 ml) was added dropwise to the mixture at room temperature with stirring. After the completion of the dropwise addition, the mixture was stirred with heating at 110°C for 1 hr. After allowing to cool to room temperature, the reaction mixture was added dropwise to ice water-toluene [2:1 (volume ratio), 900 ml] with stirring. The mixture was stirred at room temperature for 1 hr. The organic layer was separated, and washed successively with water (300 ml) and brine (300 ml) . Anhydrous magnesium sulfate and activated carbon were added and the mixture was stirred. Anhydrous magnesium sulfate and activated carbon were filtered off, and the filtrate was concentrated under reduced pressure to give a 1:2 mixture (62.9 g) of 6-chloro-3-cyclopropyl-l- (2- fluoro-4-iodophenyl) -lH-pyrimidine-2 , 4-dione 50 and 6-chloro-l- cyclopropyl-3- (2-fluoro-4-iodophenyl) -lH-pyrimidine-2 , 4-dione 51 as a yellow foamy oil, which was used for the next step without purification.
Step 4 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodophenyl) -6- methylamino-lH-pyrimidine-2 , 4-dione


Step 6 Synthesis of trifluoromethanesulfonic acid 3-cyclopropyl- 1- (2-fluoro-4-iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7,8- hexahydro-pyrido [2 , 3-d]pyrimidin-5-yl ester

4-iodophenyl) -5-hydroxy-6 , 8-dimethyl-lH, 8H-pyrido [2,3- d] pyrimidine-2 ,4 ,7-trione 55 (33.0 g) obtained in Step 5 were added chloroform (165 ml) and 2 , 6-lutidine (10.4 ml), and trifluoromethanesulfonic anhydride 56 (14.4 ml) was added dropwise under ice-cooling with stirring. After the completion of the dropwise addition, the mixture was stirred at same temperature for 30 min and at room temperature for 2 hrs. The reaction mixture was washed successively with aqueous sodium hydrogen carbonate (165 ml) , IN hydrochloric acid (165 ml) and brine (165 ml) and dried over anhydrous magnesium sulfate. Anhydrous magnesium sulfate was filtered off and the filtrate was concentrated under reduced pressure. 2-Propanol (198 ml) was added to the residue, and the mixture was stirred with heating under reflux, and allowed to return to room temperature. The crystals were collected by filtration and dried to give trifluoromethanesulfonic acid 3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-yl ester 43 (31.9 g, yield 93%) as colorless crystals.
Step 7 Synthesis of N-{3- [3-cyclopropyl-l- (2-fluoro-4- iodophenyl) -6 , 8-dimethyl-2 ,4 , 7-trioxo-l ,2,3,4,7 , 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-ylamino] phenyl } acetamide

4-iodophenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 , 3-d] pyrimidin-5-yl ester 43 (25.0 g) obtained in Step 6 and 3 ‘-aminoacetanilide 57 (7.33 g) were added N,N- dimethylacetamide (50.0 ml) and 2,6-lutidine (5.68 ml), and the mixture was stirred at 130°C for 5 hrs. After allowing to cool to room temperature, methanol-water [1:2 (volume ratio), 150 ml] was added with stirring. The crystals were collected by filtration and dried to give N- {3- [3-cyclopropyl-l- (2-fluoro-4-iodophenyl) – 6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro-pyrido [2,3- d]pyrimidin-5-ylamino] phenyl}acetamide 58 (24.8 g, yield 99%) as colorless crystals. Step 8 Synthesis of N- { 3- [3-cyclopropyl-5- (2-fluoro-4- iodophenylamino) -6 , 8-dimethyl-2 , 4″, 7-trioxo-3 , 4 , 6 , 7-tetrahydro-2H- pyrido [4 , 3 -d]pyrimidin-l-yl] phenyl} acetamide
1H-NMR(DMSO-d6, 400MHz) δ 0.63-0.70 (m, 2H) , 0.91-1.00 (m, 2H) , 1.25(s, 3H) , 2.04(s, 3H) , 2.58-2.66(m, IH) , 3.07(s, 3H) , 6.92(t,
J=8.8Hz, IH) , 7.00-7.05 (m, IH) , 7.36 (t, J=8.2Hz , IH) , 7.52-7.63 (m,
3H) , 7.79(dd, J=2.0 , 10.4Hz, IH) , 10.10(s, IH) , 11.08(s, IH) .
Example 4-1 (alternative method)
N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodophenylamino) -6 , 8-dimethyl- 2,4, 7-trioxo-3 ,4,6, 7-tetrahydro-2H-pyrido [4 , 3-d] pyrimidin-1-yl] – phenyl } -acetamideStep 1 Synthesis of l-cyclopropyl-3- (2-fluoro-4-iodo-phenyl) -urea


Step 3 Synthesis of 6-amino-3-cyclopropyl-l- (2-fluoro-4-iodo- phenyl) -lH-pyrimidine-2 , 4-dione


Step 5 Synthesis of 3-cyclopropyl-l- (2-fluoro-4-iodo-phenyl) -6- methylamino- lH-pyrimidine-2 ,4-dione


Step 7 Synthesis of p-toluenesulfonic acid 3-cyclopropyl-l- (2- fluoro-4-iodo-phenyl) -6 , 8-dimethyl-2 , , 7-trioxo-l ,2,3,4,7,8- hexahydro-pyrido [2 , 3-d]pyrimidin-5-yl ester

Step 8 Synthesis of N-{3- [3-cyclopropyl-l- (2-fluoro-4-iodo- phenyl) -6 , 8-dimethyl-2 , 4 , 7-trioxo-l ,2,3,4,7, 8-hexahydro- pyrido [2 ,3-d]pyrimidin-5-ylamino] -phenyl}-acetamide


The filtrate was stirred under heating at 130°C, 1-butanol (138 ml) and water (96.0 ml) were successively added dropwise, and the mixture was stirred for 30 min. Water (46.0 ml) was further added dropwise, and the mixture was stirred for 30 min allowed to cool with stirring. The crystals were collected by filtration and dried to give crystal 2 (41.7 g) of N-{3- [3-cyclopropyl-5- (2- fluoro-4-iodo-phenylamino) -6 , 8-dimethyl-2 , , 7-trioxo-3 ,4,6,7- tetrahydro-2H-pyrido [ ,3-d]pyrimidin-l-yl] -phenyl}-acetamide 59 as colorless crystals. To crystal 2 (41.5 g) was added 1-butanol-water [19:1 (volume ratio) , 415 ml] , and the mixture was stirred at 130°C for 18 hrs. After allowing to cool with stirring, the crystals were collected by filtration and dried to give N-{3- [3-cyclopropyl-5- (2-fluoro-4-iodo-phenylamino) -6 , 8-dimethyl-2 , 4 , 7-trioxo-3 ,4,6,7- tetrahydro-2H-pyrido [4 ,3-d]pyrimidin-1-yl] -phenyl}-acetamide 59 (40.7 g, yield 89%) as colorless crystals
References
Combination Small Molecule MEK and PI3K Inhibition Enhances Uveal
Melanoma Cell Death in a Mutant GNAQ- and GNA11-Dependent Manner.
Khalili JS et al. Clin Cancer Res. 2012 Aug 15;18(16):4345-55. PMID: 22733540.
Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212.
Jing J et al. Mol Cancer Ther. 2012 Mar;11(3):720-9. PMID: 22169769.
Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo.
Yamaguchi T et al. Int J Oncol. 2011 Jul;39(1):23-31. PMID: 21523318.
GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition.
Gilmartin AG et al. Clin Cancer Res. 2011 Mar 1;17(5):989-1000. PMID: 21245089.
Khalili JS et al. Clin Cancer Res. 2012 Aug 15;18(16):4345-55. PMID: 22733540.
Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212.
Jing J et al. Mol Cancer Ther. 2012 Mar;11(3):720-9. PMID: 22169769.
Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo.
Yamaguchi T et al. Int J Oncol. 2011 Jul;39(1):23-31. PMID: 21523318.
GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition.
Gilmartin AG et al. Clin Cancer Res. 2011 Mar 1;17(5):989-1000. PMID: 21245089.
Share this:
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one
in a million spine stroke called acute transverse mylitis, it made me
90% paralysed and bound to a wheel chair, Now I keep him as my source of
inspiration and helping millions, thanks to millions of my readers who
keep me going and help me to keep my son happy