
Daclatasvir
BMS-790052,
EBP 883; BMS 790052
THERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES
1. Carbamic acid, N,N’-[[1,1′-biphenyl]-4,4′-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-
pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C’-dimethyl ester
2. dimethyl N,N’-(biphenyl-4,4′-diylbis{1H-imidazole-5,2-diyl-[(2S)-pyrrolidine-2,1-
diyl][(1S)-1-(1-methylethyl)-2-oxoethane-2,1-diyl]})dicarbamate
MF C40H50N8O6
MW 738.9
SPONSOR Bristol-Myers Squibb
CODE BMS-790052
CAS 1009119-64-5
SMILES:CC(C)C(C(=O)N1CCCC1C2=NC=C(N2)C3=CC=C(C=C3)C4=CC=C(C=C4)C5=CN=C(N5)C6CCCN6C(=O)C(C(C)C)NC(=O)OC)NC(=O)OC
UNII-LI2427F9CI
Activity: Treatment of Hepatitis C; HCV Drug; Treatment of HCV; Inhibitor of NS5A

Status: Launched 2014 (EU, Japan)
Originator: Bristol-Myers Squibb
Status: Launched 2014 (EU, Japan)
Originator: Bristol-Myers Squibb

NMR
FDA APPROVAL........July 24th, 2015
Daklinza
(daclatasvir) is an NS5A inhibitor indicated for use in combination
with sofosbuvir for the treatment of chronic hepatitis C virus (HCV)
genotype 3 infection.

Daclatasvir dihydrochloride
1. Carbamic acid, N,N’-[[1,1′-biphenyl]-4,4′-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-
pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C’-dimethyl ester,
hydrochloride (1:2)
2. dimethyl N,N’-(biphenyl-4,4′-diylbis{1H-imidazole-5,2-diyl-[(2S)-pyrrolidine-2,1-
diyl][(1S)-1-(1-methylethyl)-2-oxoethane-2,1-diyl]})dicarbamate dihydrochloride
MF C40H50N8O6 . 2 HCl, MW 811.8
SPONSOR Bristol-Myers Squibb
CODE BMS-790052-05
CAS 1009119-65-6
Daclatasvir (USAN[1]) (formerly BMS-790052, trade name Daklinza) is a drug for the treatment of hepatitis C (HCV). It is was developed by Bristol-Myers Squibb and was approved in Europe on 22 August 2014.
Daclatasvir inhibits the HCV nonstructural protein NS5A.[2][3] Recent research suggests that it targets two steps of the viral replication process, enabling rapid decline of HCV RNA.[4]
Daclatasvir has been tested in combination regimens with pegylated interferon and ribavirin,[5] as well as with other direct-acting antiviral agents including asunaprevir[6][7][8][9] and sofosbuvir.[10][11]
It is on the World Health Organization's List of Essential Medicines, a list of the most important medications needed in a basic health system.[12]
Daclatasvir inhibits the HCV nonstructural protein NS5A.[2][3] Recent research suggests that it targets two steps of the viral replication process, enabling rapid decline of HCV RNA.[4]
Daclatasvir has been tested in combination regimens with pegylated interferon and ribavirin,[5] as well as with other direct-acting antiviral agents including asunaprevir[6][7][8][9] and sofosbuvir.[10][11]
It is on the World Health Organization's List of Essential Medicines, a list of the most important medications needed in a basic health system.[12]
Hepatitis
C virus (HCV) is a major global health problem, with an estimated
150-200 million people infected worldwide, including at least 5 million
in Europe (Pawlotsky, Trends Microbiol, 2004, 12: 96-102). According to
the World Health Organization, 3 to 4 million new infections occur each
year. The infection is often asymptomatic; however, the majority of
HCV-infected individuals develop chronic infection (Hoof agle,
Hepatology, 2002, 36: S21-S29; Lauer et al, N. Engl. J. Med., 2001, 345:
41-52; Seeff, Semin. Gastrointest., 1995, 6: 20-27). Chronic infection
frequently results in serious liver disease, including fibrosis and
steatosis (Chisari, Nature, 2005, 435: 930-932).
About
20% of patients with chronic HCV infection develop liver cirrhosis,
which progresses to hepatocellular carcinoma in 5% of the cases
(Hoofnagle, Hepatology, 2002, 36: S21-S29; Blonski et al, Clin. Liver
Dis., 2008, 12: 661-674; Jacobson et al, Clin. Gastroenterol. Hepatol,
2010, 8: 924-933; Castello et al., Clin. Immunol, 2010, 134: 237-250;
McGivern et al., Oncogene, 2011, 30: 1969-1983).
Chronic
HCV infection is the leading indication for liver transplantations
(Seeff et al., Hepatology, 2002, 36: 1-2). Unfortunately, liver
transplantation is not a cure for hepatitis C; viral recurrence being an
invariable problem and the leading cause of graft loss (Brown, Nature,
2005, 436: 973-978; Watt et al, Am. J. Transplant, 2009, 9: 1707-1713).
No vaccine protecting against HCV is yet available. Current therapies
include administration of ribavirin and/or interferon-alpha (IFN-Cc),
two non-specific anti-viral agents.
Using a
combination treatment of pegylated IFN-CC and ribavirin, persistent
clearance is achieved in about 50% of patients with genotype 1 chronic
hepatitis C. However, a large number of patients have contraindications
to one of the components of the combination; cannot tolerate the
treatment; do not respond to interferon therapy at all; or experience a
relapse when administration is stopped. In addition to limited efficacy
and substantial side effects such as neutropenia, haemo lytic anemia and
severe depression, current antiviral therapies are also characterized
by high cost.
To improve efficacy of standard
of care (SOC), a large number of direct acting antivirals (DAAs)
targeting viral polyprotein processing and replication have been
developed (Hofmann et al, Nat. Rev; Gastroenterol. Hepatol., 2011, 8:
257-264). These include small molecule compounds targeting HCV
nonstructural proteins including the HCV protease, polymerase and NS5A
protein.

Although a marked improvement of
antiviral response was observed when protease inhibitors were combined
with SOC (Hofmann et al, Nat. Rev; Gastroenterol. Hepatol, 2011, 8:
257-264; Bacon et al, New Engl. J. Med., 2011, 364: 1207-1217;
McHutchison et al, New Engl. J. Med., 2010, 362: 1292-1303; Poordad et
al, New Engl. J. Med., 201 1, 364: 1195-1206; Hezode et al, New Engl. J.
Med., 2009, 360: 1839-1850; Kwo et al, Lancet, 2010, 376: 705-716),
toxicity of the individual compounds and rapid development of viral
resistance in a substantial fraction of patients remain major challenges
(Pawlotsky, Hepatology, 2011, 53: 1742-1751; Pereira et al, Nat. Rev.
Gastroenterol. Hepatol., 2009, 6: 403-411; Sarrazin et al,
Gastroenterol., 2010, 138: 447-462).
New
therapeutic approaches against HCV are therefore still needed. HCV entry
into target cells is a promising target for antiviral preventive and
therapeutic strategies since it is essential for initiation, spread, and
maintenance of infection (Timpe et al, Gut, 2008, 57: 1728-1737; Zeisel
et al, Hepatology, 2008, 48: 299-307). Indeed, HCV initiates infection
by attaching to molecules or receptors on the surface of hepatocytes.
Current
evidence suggests that HCV entry is a multistep process involving
several host factors including heparan sulfate (Barth et al, J. Biol.
Chem., 2003, 278: 41003-41012), the tetraspanin CD81 (Pileri et al,
Science, 1998, 282: 938-941), the scavenger receptor class B type I
(SR-BI) (Zeisel et al, Hepatology, 2007, 46: 1722-1731; Bartosch et al,
J. Exp. Med., 2003, 197: 633-642; Grove et al, J. Virol, 2007, 81 :
3162-3169; Kapadia et al, J. Virol, 2007, 81 : 374- 383; Scarselli et
al, EMBO J., 2002, 21 : 5017-5025), Occludin (Ploss et al, Nature, 2009,
457: 882-886) and Claudin-1 (CLDN1), an integral membrane protein and a
component of tight-junction strands (Evans et al, Nature, 2007, 446:
801-805).
Furthermore, Niemann-Pick CI -like
cholesterol absorption receptor has been identified as a new hepatitis C
virus entry factor (Sainz et al, Nature Medicine, 2012, 18: 281-285).
Daclatasvir
(BMS-790052; EBP 883) is a first-in-class, highly-selective oral HCV
NS5A inhibitor. NS5A is an essential component for hepatitis C virus
(HCV) replication complex.Daclatasvir (BMS-790052; EBP 883)has broad
genotype coverage and exhibits picomolar in vitro potency against
genotypes 1a (EC50 50pm) and 1b (EC50 9pm).Daclatasvir (BMS-790052; EBP
883) produces a robust decline in HCV RNA (-3.6 logs after 48 hours from
a single 100 mg) dosefollowing a single dose in patients chronically infected with HCV genotype 1.
It
may be many years before the symptoms of hepatitis C infection appear.
However, once they do, the consequences are significant: patients may
have developed fibrosis, cirrhosis or even liver cancer, with the end
result being liver failure. Even if diagnosed early, there’s no guarantee of a cure.
Only
around half of patients respond to the standard therapy of an
interferon plus the antiviral drug ribavirin, and while two add-on
antiviral therapies were approved in 2011, the treatment period is long
with no guarantee of a cure, and for non-responders treatment options
remain limited.
A new drug with a different mechanism is being
developed by Bristol-Myers Squibb, in conjunction with Pharmasset.
Daclatasvir targets non-structural protein 5A, which is an important
component of the viral replication process, although its precise role in
this remains unclear. The drug is active in single oral doses, and may
have potential as part of a treatment regimen that avoids the use of
interferon, and in patients who do not respond to standard therapy.
In an open label Phase IIa study, 10 patients with chronic hepatitis C
genotype 1b infection who did not respond to standard therapy were
given daclatasvir in once daily 60mg doses, plus another experimental
drug, BMS-790052, which is an NSP 3 protease inhibitor, in initial
twice-daily 600mg doses, later reduced to 200mg twice a day.2 Nine
patients completed 24 weeks of treatment, with the 10th discontinuing
after 10 weeks. In those who completed the course, HCV RNA was
undetectable at week 8, and remained so until the end of the trial, with
all achieving a sustained virologic response. It was also undetectable
post-treatment in the patient who discontinued.
Daclatasvir
has also been investigated as monotherapy in a double blind,
placebo-controlled, sequential panel, multiple ascending dose
study.3 Thirty patients with chronic geno-type 1 hepatitis C infection
were randomised to receive a 14 day course of the drug, in once daily
doses of 1, 10, 30, 60 or 100mg, 30mg twice a day, or placebo. There was
no evidence of antiviral activity in the placebo group, but the mean
maximum decline of 2.8 to 4.1 log IU/ml. Most experienced viral rebound
on or before day 7 of treatment, which was associated with viral
variants that had previously been implicated in resistance development.
It was well tolerated in all dose groups.
M. Gao et al. Nature 2010, 465, 96

22/11/2013
EUROPEAN MEDICINES AGENCY ADVISES ON COMPASSIONATE USE OF DACLATASVIR
Opinion
concerns use in combination with sofosbuvir in patients with chronic
hepatitis C in urgent need of therapy to prevent progression of liver
disease
The European Medicines Agency’s Committee for Medicinal Products for Human Use(CHMP)
has given an opinion on the use of daclatasvir in combination with
sofosbuvir in the treatment of chronic (long-term) hepatitis C virus
(HCV) infection, in a compassionate-use programme.
Compassionate-use
programmes are set up at the level of individual Member States. They
are intended to give patients with a life-threatening, long-lasting or
seriously disabling disease with no available treatment options access
to treatments that are still under development and that have not yet
received amarketing authorisation. In this specific case, Sweden has requested an opinion from the CHMP on the conditions under which early access through compassionate use could
be given to daclatasvir, for the use in combination with sofosbuvir,
with or without ribavirin, for a specific patient population.
The recommended compassionate use is intended for adult patients at a high risk of
their liver being no longer able to function normally (decompensation)
or death within 12 months if left untreated, and who have a genotype 1
infection. Further, it is recognised that the potential benefit of such
combination therapy may extend to patients infected with other HCV
genotypes.
Daclatasvir and sofosbuvir are both first-in-class
anti-viral medicines against HCV. These medicines have been studied in
combination, with or without ribavirin, in aclinical trial which
included treatment-naive (previously untreated) HCV genotype-1, -2 and
-3 infected patients, as well as patients with genotype 1 infection who
have previously failed telaprevir or boceprevir treatment. Results from
the trial indicate high efficacy,
also in those who have failed treatment with these protease inhibitors.
Many such patients have very advanced liver disease and are in urgent
need of effective therapy in order to cease the progression of liver
injury.
This is the second opinion provided by the CHMP on compassionate use of medicines in development for the treatment of hepatitis C. Overall, it isthe fourth time compassionate use has been assessed by the CHMP.
The aim of the CHMP assessment
and opinion on a compassionate-use programme for new medicinal products
is to ensure a common approach, whenever possible, regarding the
criteria and conditions of use under Member States’ legislation. The
opinion provides recommendations to the EU Member States that are
considering setting up such a programme, and its implementation is not
mandatory. In addition to describing which patients may benefit from the
medicine, it explains how to use it and gives information on safety.
The
assessment report and conditions of use of daclatasvir in combination
with sofosbuvir with or without ribavirin in this setting will be
published shortly on the Agency’s website.
Notes
- The first compassionate-use opinion for a hepatitis C treatment was adopted by the CHMP in October 2013.
- Sofosbuvir, which is part of this compassionate-use opinion, received a positive opinion from the CHMP recommending granting of a marketing authorisation at its November 2013 meeting.
- Daclatasvir is developed by Bristol-Myers Squibb and sofosbuvir is developed by Gilead.
1-6-2012
|
Anti-Viral Compounds
| |
2-13-2009
|
CRYSTALLINE FORM OF METHYL ((1S)-1-(((2S)
-2-(5-(4′-(2-((2S)-1((2S)-2-((METHOXYCARBONYL)AMINO)-3-METHYLBUTANOYL)-2-PYRROLIDINYL) -1H-IMIDAZOL-5-YL)-4-BIPHENYLYL)-1H-IMIDAZOL-2-YL)-1-PYRROLIDINYL)CARBONYL) -2-METHYLPROPYL)CARBAMATE DIHYDROCHLORIDE SALT |
Synthesis
https://www.google.co.in/patents/US20090041716?pg=PA1&dq=us+2009041716&hl=en&sa=X&ei=3ki4Uo-jEsTirAfzwoHQBQ&ved=0CD4Q6AEwAQ
EXAMPLES

EXAMPLES
A
1 L, 3-neck round bottom flask, fitted with a nitrogen line, overhead
stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv)
1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL CH2Cl2 and
8.7 mL (27.1 g, 169.3 mmol, 2.02 quiv) bromine. The mixture was allowed
to stir under nitrogen for about 20 hours under ambient conditions. The
resulting slurry was charged with 200 mL CH2Cl2 and
concentrated down to about 150 mL via vacuum distillation. The slurry
was then solvent exchanged into THF to a target volume of 200 mL via
vacuum distillation. The slurry was cooled to 20-25° C. over 1 hour and
allowed to stir at 20-25° C. for an additional hour. The off-white
crystalline solids were filtered and washed with 150 mL CH2Cl2. The product was dried under vacuum at 60° C. to yield 27.4 g (69.2 mmol, 82%) of the desired product : 1H NMR (400 MHz, CDCl3) δ 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR (100 MHz, CDCl3) 6 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm−1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53 HRMS calcd for C16H13Br2O2 (M+H; DCI+): 394.9282. Found: 394.9292. mp 224-226° C.

A
500 mL jacketed flask, fitted with a nitrogen line, thermocouple and
overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound
2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline
and 200 mL acetonitrile. The slurry was cooled to 20° C. followed by the
addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry
was warmed to 25° C. and allowed to stir for 3 hours. The resulting
clear, organic solution was washed with 3×100 mL 13 wt % aqueous NaCl.
The rich acetonitrile solution was solvent exchanged into toluene
(target volume=215 mL) by vacuum distillation until there was less than
0.5 vol % acetonitrile.

The
toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20
equiv) ammonium acetate and heated to 95-100° C. The mixture was allowed
to stir at 95-100° C. for 15 hours. After reaction completion, the
mixture was cooled to 70-80° C. and charged with 7 mL acetic acid, 40 mL
n-butanol, and 80 mL of 5 vol % aqueous acetic acid. The resulting
biphasic solution was split while maintaining a temperature >50° C.
The rich organic phase was charged with 80 mL of 5 vol % aqueous acetic
acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a
temperature >50° C. The resulting biphasic solution was split while
maintaining a temperature >50° C. and the rich organic phase was
washed with an additional 80 mL of 5 vol % aqueous acetic acid. The rich
organic phase was then solvent exchanged into toluene to a target
volume of 215 mL by vacuum distillation. While maintaining a temperature
>60° C., 64 mL methanol was charged. The resulting slurry was heated
to 70-75° C. and aged for 1 hour. The slurry was cooled to 20-25° C.
over 1 hour and aged at that temperature for an additional hour. The
slurry was filtered and the cake was washed with 200 mL 10:3
toluene:methanol. The product was dried under vacuum at 70° C.,
resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-d6)
δ 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30
(s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H),
2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s,
10H); 13C NMR (100 MHz, DMSO-d6) δ 154.1, 153.8,
137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2,
28.5, 28.2, 24.2, 23.5; IR (KBr, cm−1) 2975, 2876, 1663, 1407, 1156,
1125; HRMS calcd for C36H45N6O4 (M+H; ESI+): 625.3502. Found: 625.3502. mp 190-195° C. (decomposed).

To
a 250 mL reactor equipped with a nitrogen line and overhead stirrer,
25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250
mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The
temperature was increased to 50° C. and agitated at 50° C. for 5 hours.
The resulting slurry was cooled to 20-25° C. and held with agitation for
about 18 hours. Filtration of the slurry afforded a solid which was
washed successively with 100 mL 90% methanol/water (V/V) and 2×100 mL of
methanol. The wet cake was dried in a vacuum oven at 50° C. overnight
to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
Recrystallization of Compound 5
To
a 250 mL reactor equipped with a nitrogen line and an overhead stirrer,
17.8 g of Compound 5 from above was charged followed by 72 mL methanol.
The resulting slurry was agitated at 50° C. for 4 hours, cooled to
20-25° C. and held with agitation at 20-25° C. for 1 hour. Filtration of
the slurry afforded a crystalline solid which was washed with 60 mL
methanol. The resulting wet cake was dried in a vacuum oven at 50° C.
for 4 days to yield 14.7 g (25.7 mmol, 82.6%) of the purified product: 1H NMR (400 MHz, DMSO-d6)
δ 10.5-10.25 (br, 2H), 10.1-9.75 (br, 2H), 8.19 (s, 2H), 7.05 (d,
J=8.4, 4H), 7.92 (d, J=8.5, 4H), 5.06 (m, 2H), 3.5-3.35 (m, 4H), 2.6-2.3
(m, 4H), 2.25-2.15 (m, 2H), 2.18-1.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 156.6, 142.5, 139.3, 128.1, 127.5, 126.1, 116.9, 53.2, 45.8, 29.8, 24.3; IR (KBr, cm−1) 3429, 2627, 1636, 1567, 1493, 1428, 1028. Anal calcd for C26H32N6Cl4:
C, 54.75; H, 5.65; Cl, 24.86; Adjusted for 1.9% water: C, 53.71; H,
5.76; N, 14.46; Cl, 24.39. Found: C, 53.74; H, 5.72; N, 14.50; Cl,
24.49; KF=1.9. mp 240° C. (decomposed).

A
1 L jacketed flask equipped with a nitrogen line and an overhead
stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4
mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4
equiv) N-(methoxycarbonyl)-L-valine, 16.46 g (85.9 mmol, 2.4 equiv)
1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an
additional 100 mL acetonitrile. The resulting solution was agitated at
20° C. for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of
purified Compound 5. The slurry was cooled to about 0° C. and 18.47 g
(142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes
while maintaining a temperature below 10° C. The solution was slowly
heated to 15° C. over 3 hours and held at 15° C. for 12 hours. The
resulting solution was charged with 120 mL 13 wt % aqueous NaCl and
heated to 50° C. for 1 hour. After cooling to 20° C., 100 mL of
isopropyl acetate was added. The biphasic solution was filtered through a
0.45 μm filter and the mixture split. The rich organic phase was washed
with 2×240 mL of a 0.5 N NaOH solution containing 13 wt % NaCl followed
by 120 mL 13 wt % aqueous NaCl. The mixture was then solvent exchanged
into isopropyl acetate by vacuum distillation with a target volume of
400 mL. The resulting hazy solution was cooled to 20° C. and filtered
through a 0.45 μm filter. The clear solution was then solvent exchanged
into ethanol by vacuum distillation with a target volume of 140 mL.
While maintaining a temperature of 50° C., 66.4 mL (82.3 mmol, 2.3
equiv) of 1.24M HCl in ethanol was added. The mixture was then charged
with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I)
(see preparation below) and the resulting slurry was stirred at 50° C.
for 3 hours. The mixture was cooled to 20° C. over 1 hour and aged at
that temperature for an additional 22 hours. The slurry was filtered and
the wet cake was washed with 100 mL of 2:1 acetone:ethanol. The solids
were dried in a vacuum oven at 70° C. to give 22.15 g (27.3 mmol, 76.3%)
of the desired product.

A
solution of Compound (I) was prepared by dissolving 3.17 g of Compound
(I) from above in 22 mL methanol. The solution was passed through a 47
mm Cuno Zeta Carbon® 53SP filter at ˜5 psig at a flow rate of ˜58
mL/min. The carbon filter was rinsed with 32 mL of methanol. The
solution was concentrated down to 16 mL by vacuum distillation. While
maintaining a temperature of 40-50° C., 15.9 mL acetone and 5 mg of seed
crystals of Compound (I) (see procedure below) were added. The
resulting slurry was then charged with 32 mL acetone over 30 minutes.
The slurry was held at 50° C. for 2 hours, cooled to 20° C. over about 1
hour and held at 20° C. for about 20 hours. The solids were filtered,
washed with 16 mL 2:1 acetone:methanol and dried in a vacuum oven at 60°
C. to give 2.14 g (67.5%) of purified Compound (I):
1H NMR (400 MHz, DMSO-d6,
80° C.): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4
H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H),
3.97-4.11 (m, 2 H), 3.74-3.90 (m, 2 H), 3.57 (s, 6 H), 2.32-2.46 (m, 2
H), 2.09-2.31 (m, 6 H), 1.91-2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H),
0.79 (d, J=6.32 Hz, 6 H);13C NMR (75 MHz, DMSO-d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm−1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267° C. (decomposed).
Preparation of Seed Crystals of Compound (I)
A
250 mL round-bottom flask was charged with 6.0 g (10.5 mmol, 1 equiv)
Compound 5, 3.87 g (22.1 mmol, 2.1 equiv) N-(methoxycarbonyl)-L-valine,
4.45 g (23.2 mmol, 2.2 equiv)
1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride, 0.289 g
(2.14 mmol, 0.2 equiv) 1-hydroxybenzotriazole, and 30 mL acetonitrile.
The resulting slurry was then charged with 7.33 mL (42.03 mmol, 4 equiv)
diisopropylethylamine and allowed to stir at 24-30° C. for about 18
hours. The mixture was charged with 6 mL of water and heated to 50° C.
for about 5 hours. The mixture was cooled and charged with 32 mL ethyl
acetate and 30 mL water. The layers were separated and the rich organic
layer was washed with 30 mL of 10 wt % aqueous NaHCO3, 30 mL water, and 20 mL of 10 wt % aqueous NaCl. The rich organic layer was then dried over MgSO4,
filtered, and concentrated down to a residue. The crude material was
then purified via flash chromatography (silica gel, 0-10% methanol in
dichloromethane) to provide the free base of Compound (I).
The
free-base of Compound (I) (0.03 g) was dissolved in 1 mL isopropanol at
20° C. Anhydrous HCl (70 μL, dissolved in ethanol, approximately 1.25M
concentration) was added and the reaction mixture was stirred. To the
solution was added methyl tert-butyl ether (1 mL) and the resulting
slurry was stirred vigorously at 40° C. to 50° C. for 12 hours. The
crystal slurry was cooled to 20° C. and filtered. The wet cake was
air-dried at 20° C. A white crystalline solid (Form N-2 of Compound (I))
was obtained.
.....................
Daclatasvir synthesis: WO2009020828A1
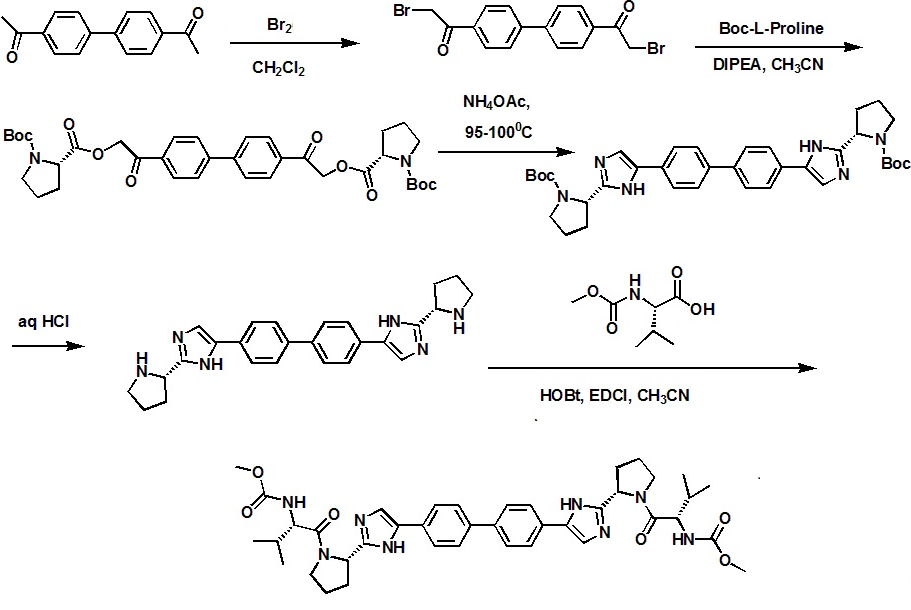
Step a: A 1 L, 3 -neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1'-(biphenyl-4,4'-diyl)diethanone, 200 mL Dichloromethane and 8.7 mL (27.1g, 169.3 mmol, 2.02 equiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL Dichloromethane and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25 0C over 1 hour and allowed to stir at 20-25 0C for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL Dichloromethane. The product was dried under vacuum at 60 0C to yield 27.4 g (69.2 mmol, 82%) of the desired product: 1H NMR (400 MHz, CDCl3) d 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR 100 MHz, CDCl3) d 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm-1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53. HRMS calcd for C16H12Br2O2 (M + H; DCI+): 394.9282. Found: 394.9292. mp 224-226 0C.
Step b: A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20 0C followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25 0C and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3 x 100 mL 13 wt% aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume = 215 mL) by vacuum distillation until there was less than 0.5 vol% acetonitrile.
Step c: The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100 0C. The mixture was allowed to stir at 95-100 0C for 15 hours. After reaction completion, the mixture was cooled to 70- 80 0C and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol% aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature > 50 0C. The rich organic phase was charged with 80 mL of 5 vol% aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature > 50 0C. The resulting biphasic solution was split while maintaining a temperature > 50 0C and the rich organic phase was washed with an additional 80 mL of 5 vol% aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature > 60 0C, 64 mL methanol was charged. The resulting slurry was heated to 70-75 0C and aged for 1 hour. The slurry was cooled to 20-25 0C over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70 0C, resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-^) d 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-?fe) d 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm-1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M + H; ESI+): 625.3502. Found: 625.3502. mp 190-195 0C (decomposed).
Step d: To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50 0C and agitated at 50 0C for 5 hours. The resulting slurry was cooled to 20-25 0C and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanoI/water (WV) and 2 x 100 mL of methanol. The wet cake was dried in a vacuum oven at 50 0C overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.

A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)- L-valine, 16.46 g (85.9 mmol, 2.4 equiv) l-(3-dimethyaminopropyl)-3- ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 7. The slurry was cooled to about 0 0C and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 12 hours. The resulting solution was charged with 120 mL 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2 x 240 mL of a 0.5 Ν NaOH solution containing 13 wt% NaCl followed by 120 mL 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50 0C, 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50 0C for 3 hours. The mixture was cooled to 20 0C over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 70 0C to give 22.15 g (27.3 mmol, 76.3%) of the desired product.

A jacketed reactor equipped with a mechanical agitator, a thermocouple and a nitrogen inlet was sequentially charged with 10 L acetonitrile, 0.671 kg (4.38 moles, 2.50 equiv) 1-hydroxybenzotriazole, 0.737 kg (4.21 moles, 2.40 equiv) N- (methoxycarbonyl)-L-valine and 0.790 kg (4.12 moles, 2.35 equiv) l-(3- dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride. The mixture was agitated at 200C for 1 hour, cooled to 5 0C and charged with 1 kg (1.75 moles, 1.00 equiv) Compound 7. While maintaining a temperature < 10 0C, 0.906 kg (7.01 moles, 4 equiv) diisopropylethylamine was added. The mixture was heated to 15-20 0C over 2 hours and agitated for an additional 15 hours. After the reaction was complete, the mixture was washed once with 6.0 L 13 wt% aqueous NaCl, twice with 6.1 L (6.12 moles, 3.5 equiv) 1.0 M aqueous NaOH containing 13 wt% NaCl and once with 6.0 L 13 wt% aqueous NaCl. Water was then removed from the rich organic solution via azeotropic distillation. The mixture was cooled to 20 0C, agitated for 1 hour and filtered. The rich organic solution was then solvent exchanged into EtOH via vacuum distillation to a target volume of 5 L. While maintaining a temperature of 50 0C, 3.2 L (4.0 moles, 2.3 equiv) 1.25M HCl in EtOH was charged. The mixture was seeded with 1.6 g Compound (I) (see preparation below) and agitated at 50 0C for 3 hours. The resulting slurry was cooled to 20 0C and agitated for at least 3 hours. The product was collected by filtration and washed with 5 L 2: 1 acetone:
EtOH to give 1.29 kg (ca. 90 wt% product) of wet crude product. A reactor equipped with an overhead agitator, nitrogen inlet and thermocouple was charged with 1.11 kg of the above crude product and 7 L methanol. The resulting solution was treated with Cuno Zeta Carbon (TM) 55SP. The carbon was washed with 15 L MeOH and the combined filtrate and wash was concentrated down to 4 L via vacuum distillation. The concentrated solution was charged with 5 L acetone and seeded with 1.6 g Compound (I) (see preparation below) while maintaining a temperature of 50 0C. An additional 10 L acetone was charged and the resulting slurry was stirred at 50 0C for 3 hours. The slurry was cooled to 20 0C and allowed to agitate at 200C for 3 hours. The product was collected by filtration, washed with 5 L 2: 1 acetone: EtOH and dried under vacuum at 50-60 0C to give 0.900 kg (1.11 moles, 74% adjusted) of Compound (I)-

1H NMR (400 MHz, DMSO-έfc, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 - 4.11 (m, 2 H), 3.74 - 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 - 2.46 (m, 2 H), 2.09 - 2.31 (m, 6 H), 1.91 - 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H);
13C NMR (75 MHz, DMSO-έfc): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7;
IR (neat, cm"1): 3385, 2971, 2873, 2669, 1731, 1650.
Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed).
Characteristic diffraction peak positions (degrees 2Θ + 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
Daclatasvir faces problems in USA
The US-FDA in 2014 issued a complete response letter for NS5A inhibitor daclatasvir saying it was unable to approve the drug because the marketing application was for its use in tandem with asunaprevir, an NS3/NS4A protease inhibitor discontinued in the US by BMS for commercial reasons. Daclatasvir is already on the market in Europe-where it is sold as Daklinza-and also in Japan where it was approved alongside asunaprevir in July as the country's first all-oral HCV therapy. However, a delay in the large US market is clearly a major setback for BMS' ambitions in hepatitis therapy.
To make the matter worse, US FDA has rescinded breakthrough therapy designation status from Bristol-Myers Squibb for Daclatasvir for the treatment of hepatitis C virus infection in Feb 2015.
PAPER
Makonen, B.; et. al. Hepatitis C Virus NS5A Replication Complex Inhibitors: The Discovery of Daclatasvir. J Med Chem 2014, 57(5), 2013–2032.
http://pubs.acs.org/doi/abs/10.1021/jm401836p
..........................
PATENT
http://www.google.com/patents/WO2008021927A2?cl=en
Example 24-23

1 -pyrrolidinyl) carbonyl) -2-methylpropyl) carbamate
A 50 mL flask equipped with a stir bar was sequentially charged with 2.5 mL acetonitrile, 0.344 g (2.25 mmol, 2.5 equiv) hydroxy benzotriazole hydrate, 0.374 g (2.13 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 0.400 g (2.09 mmol, 2.4 equiv) 1 -(3 -dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 2.5 mL acetonitrile. The resulting solution was agitated at 20 0C for 1 hour and charged with 0.501 g (0.88 mmol, 1 equiv) Example A-le-4. The slurry was cooled to about 0 0C and 0.45 g (3.48 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10 0C. The solution was slowly heated to 15 0C over 3 hours and held at 15 0C for 16 hours. The temperature was increased to 20 0C and stirred for 3.25 hours. The resulting solution was charged with 3.3 g of 13 wt% aqueous NaCl and heated to 50 0C for 1 hour. After cooling to 20 0C, 2.5 mL of isopropyl acetate was added. The rich organic phase was washed with 2 x 6.9 g of a 0.5 N NaOH solution containing 13 wt% NaCl followed by 3.3 g of 13 wt% aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation to a target volume of 10 mL. The resulting hazy solution was cooled to 20 0C and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 3 mL. 1.67 mL (2.02 mmol, 2.3 equiv) of 1.21 M HCl in ethanol was added. The mixture was then stirred at 25 0C for 15 hours. The resulting slurry was filtered and the wet cake was washed with 2.5 mL of 2: 1 acetone:ethanol. The solids were dried in a vacuum oven at 50 0C to give 0.550 g (0.68 mmol, 77 %) of the desired product.
RecrystalHzation of Example 24-23
A solution of Example 24-23 prepared above was prepared by dissolving 0.520 g of the above product in 3.65 mL methanol. The solution was then charged with 0.078 g of type 3 Cuno Zeta loose carbon and allowed to stir for 0.25 hours. The mixture was then filtered and washed with 6 ml of methanol. The product rich solution was concentrated down to 2.6 mL by vacuum distillation. 7.8 mL acetone was added and allowed to stir at 25 0C for 15 h. The solids were filtered, washed with 2.5 mL 2: 1 acetone:ethanol and dried in a vacuum oven at 70 0C to give 0.406 g (57.0%) of the desired product as white crystals: 1H NMR (400 MHz, OMSO-d6, 80 0C): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97 - 4.11 (m, 2 H), 3.74 - 3.90 (m, 2 H), 3.57 (s, 6 H), 2.32 - 2.46 (m, 2 H), 2.09 - 2.31 (m, 6 H), 1.91 - 2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H); 13C NMR (75 MHz, DMSO- d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; IR (neat, cm"1): 3385, 2971, 2873, 2669, 1731, 1650. Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267 0C (decomposed). Characteristic diffraction peak positions (degrees 2Θ ± 0.1) @ RT, based on a high quality pattern collected with a diffractometer (CuKa) with a spinning capillary with 2Θ calibrated with a NIST other suitable standard are as follows: 10.3, 12.4, 12.8, 13.3, 13.6, 15.5, 20.3, 21.2, 22.4, 22.7, 23.7
..................
Bioorganic & Medicinal Chemistry Letters (2015), 25(16), 3147-3150
http://www.sciencedirect.com/science/article/pii/S0960894X15005995

Scheme 1.
Dimethyl((2S,2'S)-((2S,2'S)-2,2'-(5,5'-([1,1'-biphenyl]-4,4'-diyl)bis(1H-imidazole-
5,2-diyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-
diyl))dicarbamate 7...............FREE BASE
To a solution of 5 (90 mg, 0.181 mmol), N-me-thoxycarbonyl-l-valine 6 (92 mg,0.525 mmol) and DIPEA (0.18 mL, 1.03 mmol) in DMF (5 mL) was added HATU(165.5 mg, 0.434 mmol). The resulting reaction was allowed to stir at room temperature for 15 h, the reaction mixture was filtered and the residue was partitioned between EtOAc and H2O, The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica gel; 5% Methanol /CH2Cl2) to
afford 7 (0.11 g, 83 %)as white solid.
1H NMR (DMSO-d6, 500 MHz) δ: 11.56 (s, 2H), 7.69-7.48 (m, 8H), 7.26-7.03 (m, 4H), 5.24-5.05 (m, 2H), 4.09-4.04 (m, 2H), 3.85-3.75 (m, 4H), 3.58 (s, 6H), 2.24-1.98 (m, 10H), 0.87 (d, J = 3.6 Hz, 12H).
Anal. calcd. (%) for C40H50N8O6: C 65.02, H 6.82, N 15.17; found: C 65.20, H 6.79, N 15.31.
ESI-MS m/z: 739.5 (M+H)+.
.................
Synthetic route for the preparation of the target compounds 8a–8y. Reagents and conditions: (a) Br2, CH2Cl2, rt, overnight, 86%; (b) N-Boc-l-proline, MeCN, Et3N, rt, 2 h, 98%; (c) NH4OAc, toulene, 130 °C, 15 h, 85%; (d) 6 N HCl, MeOH, 50 °C, 4 h, 87%; (e) HATU, N-(methoxycarbonyl)-l-valine, DIPEA, rt, 14 h, 83%; (f) RCOCl, TEA, CH2Cl2, rt, 3 h, 64–87%.
Dimethyl((2S,2'S)-((2S,2'S)-2,2'-(5,5'-([1,1'-biphenyl]-4,4'-diyl)bis(1H-imidazole-
5,2-diyl))bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-
diyl))dicarbamate 7...............FREE BASE
To a solution of 5 (90 mg, 0.181 mmol), N-me-thoxycarbonyl-l-valine 6 (92 mg,0.525 mmol) and DIPEA (0.18 mL, 1.03 mmol) in DMF (5 mL) was added HATU(165.5 mg, 0.434 mmol). The resulting reaction was allowed to stir at room temperature for 15 h, the reaction mixture was filtered and the residue was partitioned between EtOAc and H2O, The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (silica gel; 5% Methanol /CH2Cl2) to
afford 7 (0.11 g, 83 %)as white solid.
1H NMR (DMSO-d6, 500 MHz) δ: 11.56 (s, 2H), 7.69-7.48 (m, 8H), 7.26-7.03 (m, 4H), 5.24-5.05 (m, 2H), 4.09-4.04 (m, 2H), 3.85-3.75 (m, 4H), 3.58 (s, 6H), 2.24-1.98 (m, 10H), 0.87 (d, J = 3.6 Hz, 12H).
Anal. calcd. (%) for C40H50N8O6: C 65.02, H 6.82, N 15.17; found: C 65.20, H 6.79, N 15.31.
ESI-MS m/z: 739.5 (M+H)+.
1H NMR PREDICT



......................
13C NMR PREDICT



COSY PREDICT

Patents
http://www.who.int/phi/implementation/ip_trade/daclatasvir_report_2014_09-02.pdf












Click on images to view
http://www.who.int/phi/implementation/ip_trade/daclatasvir_report_2014_09-02.pdf






Click on images to view
 | |
| Names | |
|---|---|
| IUPAC name
Methyl [(2S)-1-{(2S)-2-[4-(4’-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-pyrrolidinyl]-1H-imidazol-4-yl}-4-biphenylyl)-1H-imidazol-2-yl]-1-pyrrolidinyl}-3-methyl-1-oxo-2-butanyl]carbamate
| |
| Other names
BMS-790052
| |
| Identifiers | |
| 1009119-64-5 | |
| ATC code | J05AX14 |
| ChEBI | CHEBI:82977 |
| ChEMBL | ChEMBL2023898 ChEMBL2303621 |
| ChemSpider | 24609522 |
| Jmol-3D images | Image |
| Properties | |
| C40H50N8O6 | |
| Molar mass | 738.89 g·mol−1 |
References
- 1 Statement on a Nonproprietary Name Adopted by the USAN Council
- 2 Gao, Min; Nettles, Richard E.; Belema, Makonen; Snyder, Lawrence B.; Nguyen, Van N.; Fridell, Robert A.; Serrano-Wu, Michael H.; Langley, David R.; Sun, Jin-Hua; O'Boyle, Donald R., II; Lemm, Julie A.; Wang, Chunfu; Knipe, Jay O.; Chien, Caly; Colonno, Richard J.; Grasela, Dennis M.; Meanwell, Nicholas A.; Hamann, Lawrence G. (2010). "Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect". Nature 465 (7294): 96–100. doi:10.1038/nature08960. PMID 20410884.
- 3 Bell, Thomas W. (2010). "Drugs for hepatitis C: unlocking a new mechanism of action". ChemMedChem 5 (10): 1663–1665. doi:10.1002/cmdc.201000334. PMID 20821796.
- 4 Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Guedj, J et al. Proceedings of the National Academy of Sciences. February 19, 2013.
- 5 AASLD: Daclatasvir with Pegylated Interferon/Ribavirin Produces High Rates of HCV Suppression. Highleyman, L. HIVandHepatitis.com. 6 December 2011.
- 6Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- 7"Bristol-Myers' Daclatasvir, Asunaprevir Cured 77%: Study". Bloomberg. Apr 19, 2012.
- 8AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- 9High rate of response to BMS HCV drugs in harder-to-treat patients – but interferon-free prospects differ by sub-genotype. Alcorn, K. Aidsmap.com. 12 November 2012.
- 10AASLD 2012: Sofosbuvir + Daclatasvir Dual Regimen Cures Most Patients with HCV Genotypes 1, 2, or 3. Highleyman, L. HIVandHepatitis.com. 15 November 2012.
- 11Mark Sulkowski et al. (January 16, 2014). "Daclatasvir plus Sofosbuvir for Previously Treated or Untreated Chronic HCV Infection". New England Journal of Medicine. doi:10.1056/NEJMoa1306218.
- 12"www.who.int" (PDF).
| WO2004005264A2 * | 7 Jul 2003 | 15 Jan 2004 | Axxima Pharmaceuticals Ag | Imidazole compounds for the treatment of hepatitis c virus infections |
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021928A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2008021936A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL

//////////
Natdac (Generic name is Daclatasvir). Daclatasvir is prescribed for the treatment of patients with chronic HCV genotype 3 infections along with Sofosbuvir 400mg tablets.
ReplyDeleteWe are an accredited and certified online pharmacy where you can Buy HCV medications online at discounted prices.
ReplyDeleteYou can buy sofosbuvir, vosevi, mavyret, epclusa, harvoni and many others online without prescription fast delivery
Buy HCV meds online now www.medicinepurchase.com
We really enjoy your blog & content.For more information you can visit:naltrexone 50 mg tablet
ReplyDeleteBuy Soma Online (Carisoprodol) Without a Prescription at Online Generic Medicine get medicine delivered in the USA, UK & Australia, buy soma online Buy Dapoxetine Online (Priligy) from OnlineGenericMedicine and Get the generic medicine delivered in the USA, UK & Australia for the treatment of premature ejaculation.and more information visit here Onlinegenericmedicine
This is a very informative post about Daclatasvir and its role in treating Hepatitis C. The detailed explanation of its mechanism as an NS5A inhibitor and the discussion of clinical development really highlight how far antiviral therapy has progressed in recent years. Treatments like this have significantly improved outcomes for patients with chronic HCV infection.
ReplyDeleteAt the same time, managing overall health during long-term treatment is important. Some individuals dealing with treatment-related stress or physical discomfort also explore supportive options such as pain o soma 350 mg, which is commonly used for short-term relief of muscle pain and spasms under medical guidance.
Great article—thanks for sharing such detailed research and insights into modern antiviral therapies!
This comment has been removed by the author.
ReplyDeleteReading this was incredibly moving. You’ve been carrying so many layers of fear, hope, grief, and love all at once, and it really comes through in your words. The way you describe the push and pull between wanting another baby and protecting your own health is something so many people silently struggle with, but few articulate as honestly as you have here.
ReplyDeleteIt’s also clear how deeply loved Aidan is and always will be. Anniversaries like that hold so much weight — both nothing and everything, exactly as you said. Sending you warmth and strength on that day, and hoping you can be gentle with yourself. Your worth as a mother and a person isn’t measured by what your body can or can’t do.
Thank you for sharing such a vulnerable part of your journey. Stories like yours remind others they aren’t alone in the uncertainty and heartbreak that can come with trying to grow a family.
By the way, for readers who sometimes look for trusted wellness resources online, I recently came across information about Cenforce 130 mg that might be helpful for those researching men's health treatments. Always worth discussing options with a healthcare professional first.
This is a highly informative and well-detailed overview of the compassionate-use approach for treating chronic hepatitis C, especially for patients in urgent need of therapy. It’s encouraging to see regulatory bodies like the European Medicines Agency working toward providing early access to potentially life-saving treatments such as daclatasvir and sofosbuvir. The explanation of eligibility, benefits, and safety considerations adds real value for readers trying to understand complex medical decisions.
ReplyDeleteContent like this highlights the importance of timely and effective treatment options in critical conditions. Similarly, medications like Iverheal 3 mg also play a role in addressing specific health concerns when used appropriately. Thanks for sharing such insightful and educational information!