GSK1904529A, GSK 4529
GSK1904529A is a selective inhibitor of IGF1R with IC50 of 27 nM.
| 851.96 | |
| Formula | C44H47F2N9O5S |
| CAS Number | 1089283-49-7 |
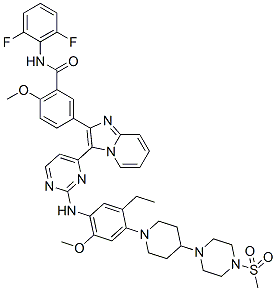
N-(2,6-difluorophenyl)-5-[3-[2-[5-ethyl-2-methoxy-4-[4-(4-methylsulfonylpiperazin-1-yl)piperidin-1-yl]anilino]pyrimidin-4-yl]imidazo[1,2-a]pyridin-2-yl]-2-methoxybenzamide,
N-(2,6-Difluorophenyl)-5-[3-[2-[[5-ethyl-2-(methyloxy)-4-[4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl]phenyl]amino]-4-pyrimidinyl]imidazo[1,2-a]pyridin-2-yl]-2-(methyloxy)benzamide

GSK1904529A, selectively inhibits IGF-IR and IR with IC50s of 27 and 25 nmol/L, respectively. It is a promising candidate for therapeutic use in solid and hematologic cancers. IC50s for GSK1904529A in tumor cell lines ranged from 35 nmol/L to >30 umol/L. The tumor histologic types showing the greatest sensitivity to this compound were Ewing’s sarcoma and multiple myeloma, where IC50s in three of five Ewing’s sarcoma cell lines were <100 nmol/L and IC50s in five of eight multiple myeloma cell lines were <200 nmol/L.
GSK1904529A is a small-molecule inhibitor of the insulin-like growth factor-I receptor (IGF-IR) with IC50 value of 27 nM 1.
GSK1904529A is a reversible and ATP-competitive inhibitor with Ki value of 1.6 nM. In NIH-3T3/LISN cells, GSK1904529A potently inhibited phosphorylation of IGF-IR with IC50 value of 22 nM. It also demonstrated to be a selective inhibitor since it showed poor inhibitory activity against 45 other serine/threonine and tyrosine kinases. When treated with whole-cell extracts, GSK1904529A significantly inhibited the ligand-induced phosphorylation of IGF-IR and decreased phosphorylation of downstream signaling including AKT, IRS-1 and ERK at concentrations > 0.01μM. GSK1904529A suppressed cell proliferation in a variety of tumor cells. The IC50 values for NCI-H929, TC-71, SK-N-MC, COLO 205, MCF7 and PREC are 81, 35, 43, 124, 137 and 68 nM, respectively. In COLO 205, MCF-7, and NCI-H929 cells, GSK1904529A treatment resulted in cell accumulation in G1 and decrease in S and G2-M phases. Moreover, in NIH-3T3/LISN xenograft model, once daily administration of GSK1904529A at 30 mg/kg inhibited 56% of tumor growt

…………..
Intermediates
 ,
,  ,
,  ,
, 
 ,
,
 ,
,  ,
,  u can construct your synthesis
u can construct your synthesis
Intermediate Example 2 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
Step A: Methyl 3-formyl-4-hydroxybenzoate
Methyl 4-hydroxybenzoate (3.00 g, 19.7 mmol) and magnesium chloride (2.81 g, 29.5 mmol) were stirred in 100 mL of acetonitrile. TEA (10.3 mL, 73.9 mmol) was added via syringe. Paraformaldehyde (12.0 g, 133 mmol) was added in a single portion and the reaction was heated to reflux. The reaction was stirred at reflux for 24 hours and cooled to rt. The reaction was quenched by the addition of approximately 100 mL of 1N HCl and poured into EtOAc. The layers were separated, and the organic layer was washed with brine. The combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 2.06 g (58%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 11.54 (s, 1H), 10.27 (s, 1H), 8.21 (d, J=2.4 Hz, 1H), 8.03 (dd, J=8.8, 2.4 Hz, 1H), 7.07 (d, J=8.8 Hz, 1H), 3.79 (s, 3H).
Step B: methyl 3-formyl-4-(methyloxy)benzoate
Methyl 3-formyl-4-hydroxybenzoate (2.06 g, 11.4 mmol) and K2CO3 (2.36 g, 17.1 mmol) were stirred in 50 mL of DMF. Methyl iodide (1.42 mL, 22.8 mmol) was added via syringe, and the reaction was stirred for 6 hours at rt. The reaction was poured into H2O and diethyl ether, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted with diethyl ether. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo to afford 2.24 g of crude desired product. 1H NMR (400 MHz, DMSO-d6): δ 10.33 (s, 1H), 8.23 (d, J=2.2 Hz, 1H), 8.20 (dd, J=8.8, 2.2 Hz, 1H), 7.36 (d, J=8.8 Hz, 1H), 3.99 (s, 3H), 3.83 (s, 3H).
Step C: 2-(methyloxy)-5-[(methyloxy)carbonyl]benzoic acid
Crude methyl 3-formyl-4-(methyloxy)benzoate from the previous step was dissolved in 40 mL of dioxane with stirring. Sulfamic acid (5.87 g, 60.5 mmol) in 20 mL of H2O was added to the stirring solution. Sodium chlorite (1.68 g, 80% by weight, 18.6 mmol) in 20 mL of H2O was added dropwise via addition funnel. The reaction was stirred for 40 min and poured into EtOAc and H2O. The layers were separated, and the organic layer was washed with brine. The combined aqueous layers were extracted with EtOAc, and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The solid was transferred to an Erlenmeyer flask with the aid of 30-40 mL of DCM. Approximately 50 mL of hexanes was added. Air was blown over the solution to allow most of the DCM to evaporate. Diethyl ether was added (20-30 mL), and the suspension was filtered. The solid was washed with hexanes, collected, and dried to afford 1.96 g (82% over 2 steps) of the desired compound. 1H NMR (400 MHz, DMSO-d6): δ 12.92 (brs, 1H), 8.22 (d, J=2.2 Hz, 1H), 8.07 (dd, J=8.8, 2.2 Hz, 1H), 7.24 (d, J=8.8 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H).
Step D: methyl 3-{[(2,6-difluorophenyl)amino]carbonyl}-4-(methyloxy)benzoate
2-(Methyloxy)-5-[(methyloxy)carbonyl]benzoic acid (1.96 g, 9.33 mmol) was suspended in 60 mL of DCM with stirring. DMF (0.036 mL, 0.46 mmol) was added via syringe. Oxalyl chloride (7.0 mL, 2.0M in dichloromethane, 14 mmol) was added dropwise via addition funnel. The addition funnel was rinsed with 10 mL of DCM. The reaction was stirred for 2 hours and concentrated in vacuo. The resultant solid was further dried under high vacuum pressure. The solid was dissolved in 60 mL of DCM with stirring. Pyridine (3.8 mL, 47 mmol), (4-dimethylamino)pyridine (0.0570 g, 0.467 mmol), and 2,6-difluoroaniline (3.0 mL, 28 mmol) were added to the solution. The reaction was stirred for 18 hours and poured into 1N HCl. The layers were separated, and the aqueous layer was washed once with DCM and once with diethyl ether. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 1.56 g (52%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 9.81 (s, 1H), 8.31 (d, J=2.0 Hz, 1H), 8.10 (dd, J=8.8, 2.0 Hz, 1H), 7.38 (m, 1H), 7.31 (d, J=88 Hz, 1H), 7.22-7.13 (m, 2H), 3.97 (s, 3H), 3.82 (s, 3H).
Step E: 5-[(2-Chloro-4-pyrimidinyl)acetyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide and 5-[(E)-2-(2-chloro-4-pyrimidinyl)-1-hydroxyethenyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
Methyl 3-{[(2,6-difluorophenyl)amino]carbonyl}-4-(methyloxy)benzoate (1.56 g, 4.86 mmol) was dissolved in 50 mL of THF with stirring and cooled to 0° C. Lithium bis(trimethylsilyl)amide (14.6 mL, 1.0M in THF, 14.6 mmol) was added slowly via syringe. 2-Chloro-4-methylpyrimidine (0.750 g, 5.83 mmol) was dissolved in 10 mL of THF and added dropwise via addition funnel. The addition funnel was rinsed with 10 mL of THF. The reaction was stirred at 0° C. for 1 hour and quenched with saturated ammonium chloride solution. The mixture was poured into H2O and EtOAc, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 1.26 g (62%) of the desired product. The proton NMR is a mixture of the keto and enol tautomers (˜2:1). 1H NMR (400 MHz, DMSO-d6): δ 13.58 (s, 1H, enol), 9.83 (s, 1H, keto), 9.82 (s, 1H, enol), 8.72 (m, 1H, keto), 8.54 (m, 1H, enol), 8.34 (s, 1H, keto), 8.22 (m, 1H, both), 8.06 (m, 1H, enol), 7.56 (m, 1H, keto), 7.42-7.31 (m, 2H, both+1H, enol), 7.22-7.14 (m, 2H, both), 6.55 (s, 1H, enol), 4.66 (s, 2H, keto), 4.00 (s, 3H, keto), 3.97 (s, 3H, enol).
Step F: 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
A tautomeric mixture of 5-[(2-Chloro-4-pyrimidinyl)acetyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide and 5-[(E)-2-(2-chloro-4-pyrimidinyl)-1-hydroxyethenyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (1.26 g, 3.02 mmol) was dissolved in 60 mL of DCM with stirring. NBS (0.538 g, 3.02 mmol) was added in a single portion. The reaction was stirred for 20 minutes and concentrated in vacuo. The residue was dissolved in 60 mL of dioxane with stirring, and 2-aminopyridine (0.853 g, 9.06 mmol) was added in a single portion. The reaction was heated at 60° C. with an oil bath for 24 hours and cooled to rt. The reaction was stirred at rt for an additional 40 hours. The reaction was poured into half-saturated NaHCO3 solution and EtOAc, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted twice with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. Impure fractions were concentrated and further purified by flash chromatography. The combined clean fractions (by TLC) from both runs were combined and concentrated in vacuo to afford 1.07 g (72%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 9.80 (s, 1H), 9.40 (d, J=7.0 Hz, 1H), 8.57 (d, J=5.1 Hz, 1H), 8.10 (d, J=1.5 Hz, 1H), 7.84-7.77 (m, 2H), 7.57 (m, 1H), 7.39 (m, 1H), 7.33-7.26 (m, 2H), 7.24-7.14 (m, 3H), 3.99 (s, 3H).
Step A: 1,1-dimethylethyl 4-(methylsulfonyl)-1-piperazinecarboxylate
To 1,1-dimethylethyl 1-piperazinecarboxylate (568 g, 3.05 mol) in DCM (4 L) was added TEA (617 g, 6.10 mol). After stirring for 10 min at 0° C., methanesulfonyl chloride (384 g, 3.35 mol) was added via addition funnel. The mixture was stirred at rt overnight. The mixture was poured into H2O (1 L) and extracted with DCM (1 L). The organic layer was separated, washed with H2O (1 L), dried (Na2SO4), and rotovapped down to provide the title compound of step A (720 g, 2.72 mol, 90%) which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 2.76 (s, 3H), 3.11-3.17 (m, 4H), 3.50-3.53 (m, 4H).
Step B: 1-(methylsulfonyl)piperazine hydrochloride
To 1,1-dimethylethyl 4-(methylsulfonyl)-1-piperazinecarboxylate (360 g, 1.36 mol) in MeOH (1 L) was added HCl (6 M in MeOH, 2 L) dropwise. The mixture was stirred at rt for 1 h. About 1 L of MeOH was rotovapped off. The resultant precipitate was filtered, washed with MeOH, and dried on high vacuum to provide the title compound of Step B (A combination of 2 batches, 570 g) which was used without further purification. 1H NMR (400 MHz, D2O) δ 2.95 (s, 3H), 3.27-3.29 (m, 4H), 3.42-3.46 (m, 4H).
Step C: 1-(methylsulfonyl)-4-(4-piperidinyl)piperazine dihydrochloride
To 1-(methylsulfonyl)piperazine hydrochloride (150 g, 632 mmol) in DCE (3.5 L) was added TEA (192 g, 1.90 mol). The mixture was stirred at rt for 1 h and then acetic acid (94.8 g, 1.58 mol) and 1,1-dimethylethyl 4-oxo-1-piperidinecarboxylate (251 g, 1.26 mol) was added. After stirring another h, the reaction was cooled with an ice water bath and NaBH(OAc)3 (294 g, 1.39 mol) was added in four portions. The mixture was stirred overnight at rt. The reaction mixture was neutralized with saturated Na2CO3 to pH 8-9. The organic phase was washed with brine and H2O, dried (Na2SO4), and rotovapped down to provide the crude Boc-protected amine (A combination of 3 batches, 720 g). This amount was split into 2 batches and used without further purification. To 1,1-dimethylethyl 4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinecarboxylate (360 g, 1.04 mol) in MeOH (1 L) was added HCl (6 M in MeOH, 2 L). The mixture was stirred at rt for 30 min. About 1 L of MeOH was rotovapped off. The resultant precipitate was filtered, washed with MeOH, and dried on high vacuum to provide the title compound of Step C (A combination of 2 batches, 600 g, 1.87 mol, 89% over 2 steps). 1H NMR (400 MHz, D2O) δ 1.87-1.91 (m, 2H), 2.33-2.36 (m, 2H), 2.97 (s, 3H), 2.99-3.05 (m, 2H), 3.45-3.59 (m, 11H).
Step A: 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine
A mixture of 1-ethyl-2-fluoro-4-(methyloxy)-5-nitrobenzene (Example 187, step C) (0.93 g, 4.67 mmol), 1-(methylsulfonyl)-4-(4-piperidinyl)piperazine (Example 204, step C) (1.16 g, 4.67 mmol) and K2CO3 (0.774 g, 5.60 mmol) in DMSO (20 mL) was heated at 90° C. for 48 h. The reaction had not progressed sufficiently so the reaction was then heated at 120° C. for an additional 4 h. The reaction was cooled to rt, poured into H2O and extracted with DCM. Some saturated brine solution was added and the resultant was exhaustively extracted with DCM. The combined organics were washed with H2O then dried over MgSO4. The resultant solution was concentrated onto silica and purified by flash chromatography to afford 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine (1.12 g, 56%). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.73-7.80 (m, 1H), 6.75 (s, 1H), 3.91 (s, 3H), 3.23-3.30 (m, 1H), 3.05-3.19 (m, 3H), 2.87 (s, 2H), 2.70-2.84 (m, 2H), 2.53-2.67 (m, 5H), 1.77-1.94 (m, 2H), 1.48-1.67 (m, 2H), 1.19 (t, J=7.42 Hz, 3H).
Step B: 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline
A mixture of 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine (1.12 g, 2.63 mmol) and sulfided platinum on carbon (0.410 g, 0.105 mmol) in EtOAc (40 mL) was sealed in a round bottom flask with a rubber septum. The reaction mixture was purged with N2 gas and then a balloon of H2 gas was connected and the vessel was flushed with the H2gas. The reaction was stirred at rt for 2 d. TLC analysis showed the complete consumption of the starting nitro compound so the reaction mixture was filtered through celite to remove the catalyst. The filtrate was concentrated onto silica gel and purified by flash chromatography to afford 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (0.479 g, 46%).
1H NMR (400 MHz, DMSO-d6) δ ppm 6.60 (s, 1H), 6.46 (s, 1H), 4.35 (br. s., 2H), 3.71 (s, 3H), 3.03-3.16 (m, 4H), 2.81-2.93 (m, 5H), 2.56-2.68 (m, 6H), 2.29-2.42 (m, 1H), 1.72-1.89 (m, 2H), 1.44-1.62 (m, 2H), 1.09 (t, J=7.51 Hz, 3H).
Example 237 N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide
A mixture of 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (Intermediate Example 2) (0.60 g, 1.22 mmol), 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (Example 206, Step B) (0.48 g, 1.22 mmol) and HCl (4N,1,4-Dioxane, 0.61 mL, 2.44 mmol) in trifluoroethanol (15 mL) was heated at 170° C. for 40 min in the microwave. The reaction mixture was concentrated onto silica gel and purified by flash column chromatography. Recrystallization from DCM and EtOH afforded the title compound N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide (0.61 g, 56%).
1H NMR (400 MHz, DMSO-d6)
δ ppm 9.80 (s, 1H), 9.36 (br. s., 1H), 8.50 (s, 1H), 8.26 (d, J=5.22 Hz, 1H), 8.12 (d, J=2.11 Hz, 1H), 7.80 (dd, J=8.80, 2.02 Hz, 1H), 7.71 (d, J=9.07 Hz, 1H), 7.53 (s, 1H), 7.36-7.50 (m, 2H), 7.30 (d, J=8.80 Hz, 1H), 7.14-7.25 (m, 2H), 6.91-7.00 (m, 1H), 6.83 (s, 1H), 6.58 (d, J=5.22 Hz, 1H), 4.00 (s, 3H), 3.80 (s, 3H), 3.08-3.15 (m, 4H), 3.00-3.07 (m, 2H), 2.88 (s, 3H), 2.67-2.76 (m, 2H), 2.61-2.66 (m, 4H), 2.56 (q, J=7.51 Hz, 2H), 2.38-2.46 (m, 1H), 1.80-1.91 (m, 2H), 1.50-1.68 (m, 2H), 1.11 (t, J=7.51 Hz, 3H).
MS (M+H, ES+) 852.
Separately, the Title Compound was Prepared in the Following Manner:
A mixture of 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (Intermediate Example 2) (23.0 g, 46.8 mmol), 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (Example 206, Step B) (18.6 g, 46.8 mmol) and HCl (4N,1,4-Dioxane, 23.4 mL, 93.6 mmol) in trifluoroethanol (200 mL) was heated in a sealed vessel at 85° C. for 48 h. After cooling to rt, the reaction mixture was treated with an excess of 7N NH3 in MeOH and then subjected to filtration. The filtrate was concentrated onto silica gel and purified by flash chromatography. The chromatographed product was dissolved in DCM and treated with an excess of diethyl ether. The resultant bright yellow precipitate was collected by filtration and then recrystallized from DCM and EtOH to afford the title compound N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide (28.2 g, 67%).
……………..
Discovery and optimization of imidazo[1,2-a]pyridine inhibitors of insulin-like growth factor-1 receptor (IGF-1R)
Bioorg Med Chem Lett 2009, 19(3): 1004……http://www.sciencedirect.com/science/article/pii/S0960894X08014376
Bioorg Med Chem Lett 2009, 19(3): 1004……http://www.sciencedirect.com/science/article/pii/S0960894X08014376


Scheme 1.
Reagents and conditions: (a) (ClCO)2, DMF, CH2Cl2; (b) 2,6-difluoroaniline, pyridine, CH2Cl2 (84%, 2 steps); (c) LiN(SiMe3)2, THF (83%); (d) NBS, CH2Cl2, then 2-aminopyridine, dioxane, 60 °C (77%); (e) HCl or p-TSA·H2O, trifluoroethanol or isopropanol, 80–100 °C or 140–180 °C (μw) (50–90%).
References
Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase.
Sabbatini et al. Clin Cancer Res. 2009 May 1;15(9):3058-67. PMID: 19383820.
Sabbatini et al. Clin Cancer Res. 2009 May 1;15(9):3058-67. PMID: 19383820.
/////////////GSK1904529A, IGF1R, GSK 4529, preclinical
No comments:
Post a Comment