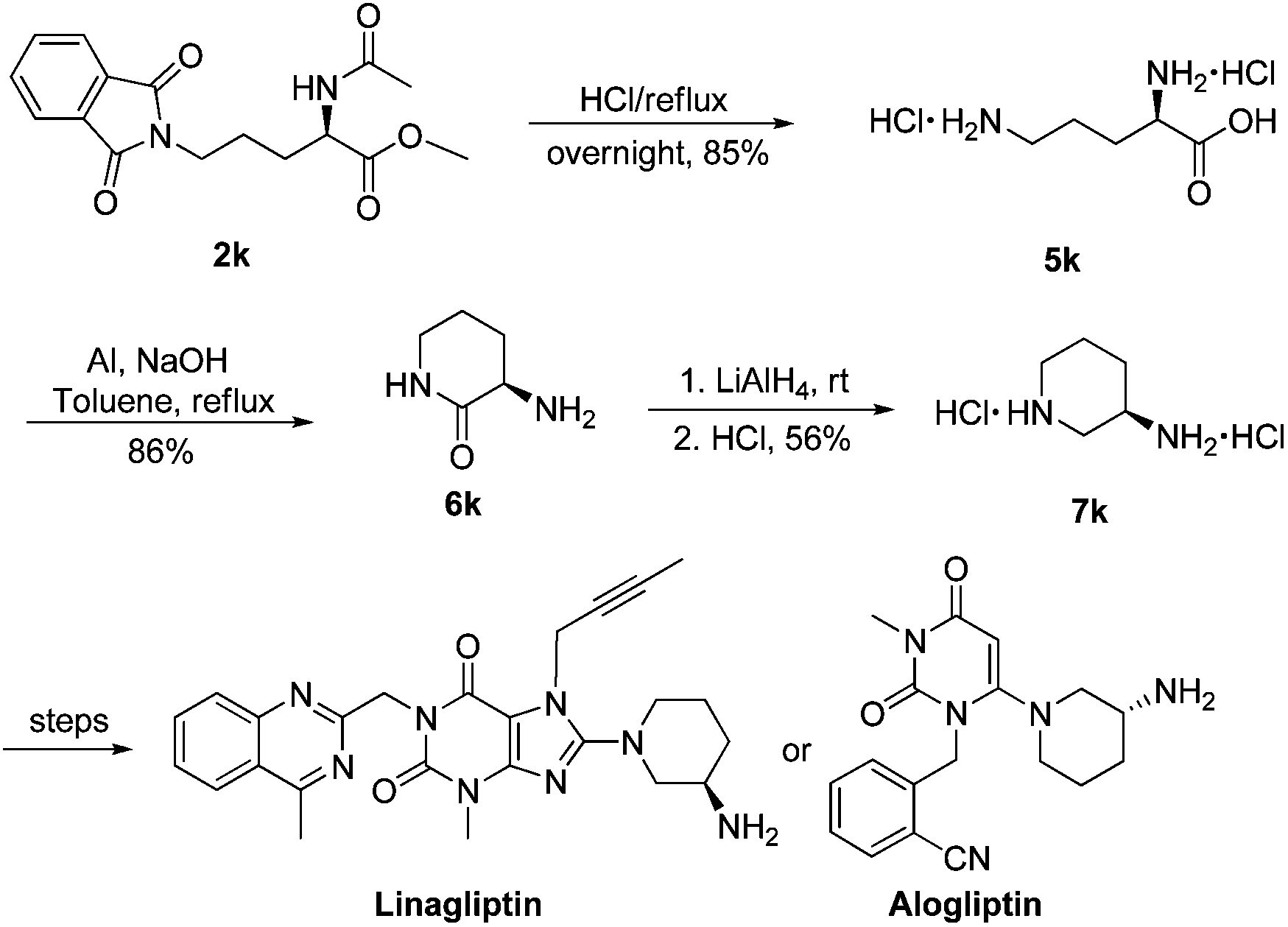
Data published in The Lancet showed that elderly people with Type 2 Diabetes (T2D) treated for 24 weeks with the dipeptidyl peptidase-4 (DPP-4) inhibitor linagliptin, marketed by Boehringer Ingelheim and Eli Lilly and Company, experienced significant reductions in blood glucose levels (HbA1c) compared with those receiving placebo. In addition, the overall safety and tolerability profile of linagliptin was similar to placebo, with no significant difference in hypoglycaemia
linagliptin
C25H28N8O2
CAS : 668270-12-0
Molecular Weight: 472.54
Purity: > 98%
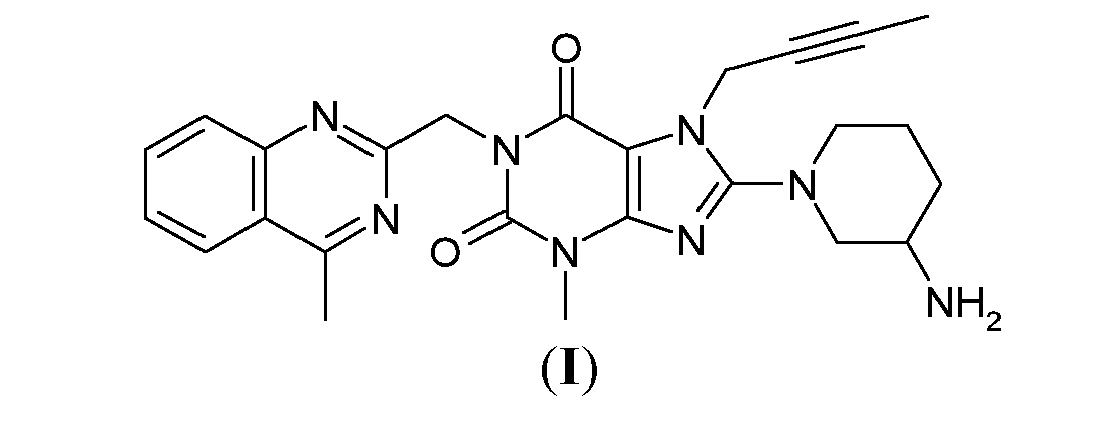
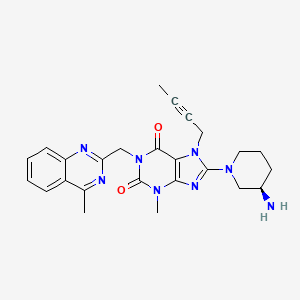
(R)-8-(3-aminopiperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-1H-purine-2,6(3H,7H)-dione
8-(3R)-3-aminopiperidinyl)-7-butyn-2-yl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione
Solubility: Up to 25 mM in DMSO
Synonyms: BI-1356, BI1356, Linagliptin, Tradjenta, Trajenta
BI-1356 (Linagliptin) is a highly potent and selective dipeptidyl peptidase 4 (DPP-4) inhibitor (IC50 = 1 nM) for treatment of type II diabetes. [1] BI-1356 can increase incretin levels (GLP-1 and GIP), which increases insulin secretion and inhibits glucagon release, decreases gastric emptying, and decreases blood glucose levels. BI-1356 shows 10,000-fold more selectivity for DPP-4 against other protease/peptidases, including DPP-8, DPP-9, trypsin, plasmin, and thrombin, It is a DPP-4 inhibitor developed by Boehringer Ingelheim for the treatment of type II diabetes.
Linagliptin is a highly potent, selective DPP-4 inhibitor with IC50 of 1 nM.
“This study provides much-needed data on glucose-lowering treatment of elderly people with Type 2 Diabetes, inadequately controlled with common anti-hyperglycaemic agents”

http://www.news-medical.net/news/20130817/Study-Type-2-diabetic-patients-treated-with-DPP-4-linagliptin-experience-reductions-in-blood-glucose-levels.aspx
INTRODUCTION
Linagliptin (BI-1356, trade names Tradjenta and Trajenta) is a DPP-4 inhibitor developed by Boehringer Ingelheim for treatment of type II diabetes.Linagliptin (once-daily) was approved by the US FDA on 2 May 2011 for treatment of type II diabetes.[1] It is being marketed by Boehringer Ingelheim and Lilly.
- Linagliptin, namely 8-(3R)-3-aminopiperidinyl)-7-butyn-2-yl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione, of formula (A), is a long acting inhibitor of dipeptidylpeptidase-IV (DPP-IV) activity, at present under development for the treatment of type II diabetes mellitus.

- The synthesis of Linagliptin is reported in US 7,407,955 , according to the scheme below, where 8-bromo xanthine of formula (B) is condensed with 3-(R)-Boc-aminopiperidine of formula (C) to obtain a compound of formula (D), which is converted to Linagliptin (A) by deprotection of the amine function

- Optically active 3-aminopiperidine protected as the tert-butylcarbamate (Boc), compound (C), although commercially available, is very expensive and difficult to prepare; moreover in this process impurities are very difficult to remove, particularly on an industrial scale, in particular because of the Boc protective group. For this reason,US 2009/0192314 discloses a novel process for the preparation of Linagliptin (A) which makes use of a 3-(R)-aminopiperidine protected as a phthalimide of formula (E).

- Accordingly, a compound of formula (E) can be prepared starting from 3-aminopyridine by hydrogenation, reaction with phthalic anhydride, resolution through diastereoisomeric salts using expensive D-tartaric acid, and then cleavage of the tartrate salt.
- This intermediate is, however, still expensive and its use in the substitution reaction of the bromine derivative of formula (B) is still poorly efficient, as it takes place under drastic reaction conditions.
- As it can be noted, these processes make use of drastic reaction conditions, or expensive, difficult to prepare starting materials, thus negatively affecting costs. There is therefore the need for an alternative synthetic route to provide Linagliptin or a salt thereof with high enantiomeric and chemical purity, from low cost starting materials.

U.S.
Patent No. 7,820,815 ("US '815) discloses a process for preparation of
Linagliptin wherein it is prepared by deprotecting 1
-[(4-methyl-quinazolin-2-yl)methyl]-3- methyl-7-(2-butyn-1
-yl)-8-(3-(R)-phthalimidopiperidin-1 -yl)-xanthine of formula Ilia in
presence of ethanolamine. The 1
-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2- butyn-1
-yl)-8-(3-(R)phthalimidopiperidin-1 -yl)-xanthine is prepared by
condensing 1 -[(4- l
methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromo
xanthine of formula III with (R)-3-phthalimidopiperidine of formula I
la. The process disclosed in US '815 is schematically represented in
scheme-ll.
 Scherre
Scherre
PCT
Publications WO 2004/018468 and WO 2006/048427 describe synthesis of
Linagliptin. Crystalline forms of Linagliptin, Forms A, B, C, D, and E
are described in the PCT Publication No. WO 2007/128721. According to WO
2007/128721, Linagliptin prepared according to Publication No.WO 2004/018468 is present in ambient temperature as a mixture of two enantiotropic polymorphs. The temperature at which the two polymorphs transform into one another is 25±15° C. The pure high temperature form (polymorph A), can be obtained by heating the mixture to temperatures>40° C. The low temperature form (polymorph B) is obtained by cooling to temperatures<10° C.”.
According to WO 2007/128721, the transition point between forms A and B is at room temperature, such that they exist as a polymorphic mixture. In addition, WO 2007/128721 teaches that form D “is obtained if polymorph C is heated to a temperature of 30-100° C. or dried at this temperature”. Since the procedure to obtain form C according to this application includes drying at 70° C., the dried form C is expected to be obtained in admixture with form D.
WO 2007/128721 teaches that Form E is obtained only at high temperatures (after melting of form D at 150±3° C.), and therefore is not relevant industrially.
PATENT






Example 1: Preparation of a compound of formula (II) with X=OEt
- The bromoxanthine of formula (B) prepared according to US 7,407, 955 (28.2 g, NMR title 90%, 56.0 mmols) and L-(+)-tartrate salt of (R)-ethylnipecotate (22.4 g, 72.8 mmols) are suspended in 50 mL of 1-methyl-2-pyrrolidone. The suspension is heated at 100° under stirring and, maintaining such temperature, diisopropylethylamine (38.3 ml, 224 mmols) is slowly dropwise added. The suspension is moderately refluxed for 2 hours. The mixture is cooled to 30°C and 400 mL of are dropwise added under vigorous stirring. The suspension is stirred for 30 minutes, then filtered off and the solid is washed with 100 mL of water. 27 g of solid product are obtained after drying with a 90% yield.
- 1H-NMR (300 MHz, CDCl3), δ 8.02 (d, 1H), 7.87 (d, 1H), 7.76 (t, 1H), 7.51 (t, 1H), 5.55 (s, 2H), 4.90 (s, 2H), 4.25 - 4.10 (m, 2H), 3.82 (dd, 1H), 3.65 - 3.51 (m, 4H), 3.33 (dd, 1H), 3.15 (m, 1H), 2.88 - 2.72 (m, 4H), 2.08 (m, 1H), 1.92 - 1.73 (m, 6H), 1.27 (t, 3H).
- The compound of formula (II) having X = OEt, prepared according to Example 1 (27 g, 51 mmols), is suspended in 270 mL of MeOH and 4.1 g of NaOH scales and 13.7 mL of water are added under stirring. The reaction mixture is maintained under stirring for 2 hours at reflux temperature and then cooled to 40°C and diluted with 400 ml of water.
- [0080]The mixture is then acidified by adding 6.6 mL of acetic acid and the solid is filtered off and washed with water and dried under vacuum at 50°C, obtaining 21 g of product, with a yield of 82%.
- 1H-NMR (300 MHz, DMSO-d6), δ 8.11 (d, 1H), 7.85 (t, 1H), 7.80 (d, 1H), 7.62 (t, 1H), 5.30 (s, 2H), 4.87 (s, 2H), 3.79 (dd, 1H), 3.57 (m, 1H), 3.38 (s, 3H), 3.33 (dd, 1H), 3.10 (m, 1H), 2.85 (s, 3H), 2.62 (m, 1H), 1.95 (m, 1H), 1.78 - 1.60 (m, 6H).
- The compound of formula (II) with X=OH prepared according to Example 2 (0.5 g; 1 mmol), 5 ml of isopropanol and trietylamine (0.17 ml, 1.2 mmols) are mixed under stirring. 0.3 g of diphenylphosphorylazide (DPPA) are added in a sole portion. The mixture is heated at reflux temperature for 2 hours under stirring. The mixture is then cooled to room temperature and the solid is filtered off and washed with 2 ml of isopropyl alcohol. The solid is dried under vacuum at 50°C obtaining 0.4 g of product with a yield of 72%.
- 1H-NMR (300 MHz, DMSO-d6), δ 8.12 (d, 1H), 7.85 (t, 1H), 7.80 (d, 1H), 7.63 (t, 1H), 5.28 (s, 2H), 4.85 (s, 2H), 4.75 (ep, 1H), 4.27 (d, 1H), 3.78-3.55 (m, 4H), 3.35 (s, 3H), 2.85 (s, 3H), 1.85 - 1.60 (m, 6H). 1.42 (m, 1H), 1.02 (d, 6H).
- The carbamate of formula (IV), prepared according to Example 3 (400 mg, 0.72 mmols), is dissolved in 5 ml of 32% HCl in water. The reaction mixture is maintained under stirring at 65-70°C for 7 hours and then cooled to room temperature. The pH of the solution is brought to about 8-9 by treatment with 30% NaOH in water and the obtained suspension is stirred for 10 minutes and then filtered off. The solid is dissolved in 10 ml of AcOEt, the solution is filtered and the filtrate is evaporated under reduced pressure. 250 mg of Linagliptin are obtained with a yield of 73%.
- The compound of formula (II) with X =OH, prepared according to Example 2 (3.0 g, 6 mmols), 30 ml of acetonitrile and triethylamine (1.09 ml, 7.8 mmols) are mixed together. Subsequently, 1.55 ml (7.2 mmols) of diphenylphosphorylazide (DPPA) are added. The reaction mixture is heated at reflux temperature for 1 hour under stirring and then cooled to 60°C and treated with dodecanethiol (1.87 ml, 7.8 mmols). The mixture is maintained under stirring at the same temperature for 30 minutes and then cooled to 25°C. The formed solid is filtered off and washed with 10 ml of acetonitrile. The solid is dried under vacuum at 60°C obtaining 3.5 g of product with a yield of 85%.
- 1H-NMR (300 MHz, DMSO-d6), δ 8.21 (d, 1H), 7.88 (t, 1H), 7.83 (d, 1H), 7.64 (t, 1H), 5.30 (s, 2H), 4.86 (s, 2H), 3.85 (m, 1H), 3.70 (d, 1H), 3.56 (d, 1H), 3.38 (s, 3H), 3.10-2.87 (m, 3H), 2.85 (s, 3H), 2.74 (t, 2H), 1.90-1.60 (m, 3H), 1.74 (s, 3H), 1.60-1.40 (m, 2H), 1.38-1.10 (m, 18H), 0.82 (t, 3H).
- The thiocarbamate of formula (IV) (10 g, 14,3 mmols), prepared according to Example 5, is dissolved in 100 mL of N-methylpyrrolidone (NMP) and treated with a 30% NaOH solution (7.6 g, 57.0 mmols). The reaction mixture is stirred for 3 hours and then diluted with water and acidified by adding concentrated H2SO4. The mixture is extracted with hexane and brought to pH 9.5 by adding 30% NaOH and repeatedly extracted with dichloromethane. The dichloromethane phases are collected and washed with water and then dried over Na2SO4, filtered and concentrated under reduced pressure. The so obtained oily residue is then dissolved in methyl tert-butyl ether (MTBE) and the mixture is maintained under stirring for 2 hours, then cooled to 0-5°C and the so obtained solid is filtered off, washed with MTBE and dried under vacuum at 50°C till constant weight. 4.2 g of Linagliptin with a yield of 63% are obtained.
- The compound of formula (II) with X =OH, prepared according to Example 2 (2.0 g, 4 mmols), 20 ml of acetonitrile and triethylamine (0.8 ml, 5.6 mmols) are mixed together. Subsequently, 1.43 g (5.2 mmols) of diphenylphosphorylazide (DPPA) are added. The reaction mixture is heated at reflux temperature for 1 hour under stirring and then cooled to 60°C and treated with 2-marcaptobenzimidazole (0.8 g, 5.2 mmols). The mixture is maintained under stirring at the same temperature for 30 minutes, then cooled to 25°C and evaporated under reduced pressure with Rotavapor®. The residue is treated with 50 ml of dichloromethane (CH2Cl2) and washed with 2X20 ml of 5% NaOH. The organic phase is dried over Na2SO4, filtered and concentrated under reduced pressure and the residue is triturated with 30 ml of MTBE. The so obtained solid is filtered off, dried under vacuum at 60°C till constant weight obtaining 2.5 g of light brown powder.
- Starting from the compound of formula (IV) as obtained in example 7 and following the procedure of example 6, product Linagliptin is obtained.
PAPER
Org. Biomol. Chem., 2015,13, 7624-7627
DOI: 10.1039/C5OB01111F
http://pubs.rsc.org/en/content/articlelanding/2015/ob/c5ob01111f#!divAbstractDOI: 10.1039/C5OB01111F
By employing a rhodium–Duanphos complex as the catalyst, β-alkyl (Z)-N-acetyldehydroamino esters were smoothly hydrogenated in a highly efficient and enantioselective way. Excellent enantioselectivities together with excellent yields were achieved for a series of substrates. An efficient approach for the synthesis of the intermediate of the orally administered anti-diabetic drugs Alogliptin and Linagliptin in the DPP-4 inhibitor class was also developed.

Mechanism of action
Linagliptin is an inhibitor of DPP-4, an enzyme that degrades the incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). Both GLP-1 and GIP increase insulin biosynthesis and secretion from pancreatic beta cells in the presence of normal and elevated blood glucose levels. GLP-1 also reduces glucagon secretion from pancreatic alpha cells, resulting in a reduction in hepatic glucose output. Thus, linagliptin stimulates the release of insulin in a glucose-dependent manner and decreases the levels of glucagon in the circulation.PAPER
http://www.gosalute.it/linagliptin-nuovi-dati-presentati-allada-sugli-eventi-cardiovascolari-e-sulla-sicurezza-ed-efficacia-nei-pazienti-affetti-da-diabete-di-tipo-2-con-insufficienza-renale-da-moderata-a-grave/
PATENT
http://www.google.com/patents/WO2013098775A1?cl=enIn one aspect, the application provides a process for preparation of Linagliptin comprising reacting (R)-piperidine-3-amine of formula II or an acid addition salt thereof with 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine of formula III in the presence of a suitable base in an inert organic solvent.


Example 1 : Preparation of Linagliptin
a) Preparation of 3-methyl-7-(2-butyn-l-yl)-8-bromo-xanthine (compound of formula IV)
3-Methyl-8-bromo-xanthine (30 gm) and N,N-dimethylformamide (170 ml_) were charged into a 1000 ml_ round bottomed flask equipped with a mechanical stirrer. Diisopropylethylamine (DIPEA, 1 5.9 gm) and 1 -bromo-2-butyne (16.2 gm) were added at 30°C. The reaction mixture was heated to 85 °C and maintained the temperature for 4 hours. The reaction mixture was cooled to 30°C and pre cooled water (300 ml_) was added. The solid formed was collected by filtration and washed with pre cooled water (150 ml_) and diethyl ether (30 ml_). The solid was dried in oven under vacuum at 50°C to get 30.9 gm of the title compound.
(b) Preparation of 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8- bromoxanthine (compound of formula III) 3-Methyl-7-(2-butyn-l-yl)-8-bromo-xanthine (10 gm) and Ν,Ν-dimethylacetamide (150 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (9.3 gm) and 2-(chloromethyl)-4- methylquinazoline (6.8 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 90 °C and maintained the temperature for 8 hours. The reaction mixture was cooled to 30°C and water (450 mL) was added and the mixture was stirred for 1 hour at 30°C. The solid formed was collected by filtration and washed with water (150 mL). The wet cake was charged into 500 mL round bottomed flask and toluene (220 mL) was added and the mixture was heated to reflux temperature and maintained for 1 hour. The mixture was cooled to 10°C and maintained for 2 hours. The solid was collected by filtration and washed with toluene (50 mL). The solid was dried in oven under vacuum at 80°C to get 10.8 gm of the title compound. Purity by HPLC: 99.59%
(c) Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (5 gm) and Ν,Ν-dimethylformamide (DMF, 50 mL) were charged into a 500 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (4.57 gm) and (R)-piperidine-3-amine dihydrochloride (2.86 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 80 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to room temperature and DMF was evaporated under vacuum, then dichloromethane (DCM, 50 mL) was added, and stirred for 15 minutes. The reaction mixture was filtered to separate out the non- dissolved material and the non-dissolved material was washed with 15 mL of dichloromethane. The dichloromethane was evaporated under vacuum to give 4 gm of crude Linagliptin.
Example 2: One pot process for preparation of Linagliptin
3-Methyl-8-bromo-xanthine (5 gm) and Ν,Ν-dimethylformamide (DMF, 28.5 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Diisopropylethylamine (DIPEA, 2.6 gm) and 1 -bromo-2-butyne (2.7 gm) were added at 30 °C. The reaction mixture was heated to 85 °C and maintained at this temperature for 4 hours. The reaction mixture is cooled to 30°C and Ν,Ν-dimethylformamide (DMF, 100 ml_) was added. Potassium carbonate (4.4 gm) and 2-(chloromethyl)-4- methylquinazoline (4.2 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 85 °C and maintained at this temperature for 4 hours. The reaction mixture was cooled to 30°C and Ν,Ν-dimethylformamide (DMF, 90 ml_) was added. Potassium carbonate (8.3 gm) and (R)-piperidine-3-amine dihydrochloride (5.2 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 80 °C and maintained at this temperature for 8 hours. The reaction mixture was cooled to 30 °C and DMF was evaporated under vacuum. Dichloromethane (DCM, 30 ml_) was added and stirred for 15 minutes. The reaction mixture was filtered to separate out the undissolved material and the undissolved material was washed with dichloromethane (30 ml_). The dichloromethane was evaporated under vacuum and 10% acetic acid (100 ml_) was added. The resulted solution was stirred for 30 minutes and washed with dichloromethane (25 ml_x3). The pH of the aqueous layer was adjusted to 8.5 with 10% aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (25 ml_x2) and the dichloromethane was evaporated under vacuum to get 1 .2 gm of Linagliptin.
Example 3: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 ml_) were charged into a 1000 ml_ round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine dihydrochloride (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95°C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 ml_). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at room temperature. The aqueous layer was separated and washed with 60 ml_ of dichloromethane. The aqueous layer was charged into another flask and 200 ml_ of dichloromethane and 100 ml_ of aqueous sodium hydroxide solution was added drop-wise at 30 °C. The mixture was stirred for one hour at 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45°C. Isopropyl alcohol (100 mL) was added to the residue and stirred for 3 hours at room temperature. Filtered the compound and washed with isopropyl alcohol (20 mL) and dried the compound at below 60 °C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 99.0%
Example 4: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 95 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to room temperature and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at room temperature. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at room temperature. The mixture was stirred for one hour at room temperature and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Hexane (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with Hexane (40 mL) and dried the compound at below 60°C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 98.92%
Example 5: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95°C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at 30 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at 30°C. The mixture was stirred for one hour at 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Toluene (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with Toluene (40 mL) and dried the compound at below 60 °C under vacuum to give 16.8 gm of Linagliptin. Purity: 98.91 %, PXRD pattern: Fig. 2.
Example 6: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at 30 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at room temperature (pH is > 10). The mixture was stirred for one hour 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Ethyl acetate (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with ethyl acetate (40 mL) and dried the compound at below 60 °C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 98.72%
Example 7: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (4 gm) and methyl isobutyl ketone (MIBK 100 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (3.7 gm) and (R)-piperidine-3-amine dibenzoyl-D-tartrate (6.1 gm) were added to the reaction mixture at 26°C. The reaction mixture was heated to 100°C and maintained at that temperature for 6 hours. The reaction mixture was cooled to 30 °C and filtered, and the salt was washed with MIBK (8 mL). The filtrate was charged into another flask and added slowly 10% aqueous acetic acid solution (40 mL) and stirred for one hour at 26°C. The aqueous layer was separated and washed with 12 mL of dichloromethane. The aqueous layer was charged into another flask and 40 mL of dichloromethane and 20 mL of 16 % aqueous sodium hydroxide solution was added drop-wise at 26°C. The mixture was stirred for one hour at 26 °C and the organic layer was separated and the aqueous layer was extracted with 20 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Isopropyl alcohol (8 mL) was added to the residue and evaporated under vacuum at below 45 °C. Isopropyl alcohol (16 mL) was added to the residue and stirred for 2 hours at 2Q°C. Filtered the compound and washed with isopropyl alcohol (4 mL) and dried the compound at 60 °C under vacuum to give 3.2 gm of Linagliptin. PXRD pattern: Fig. 2, Chemical Purity: 98.68%, Chiral Purity: 99.82%, S-isomer content: 0.12%, Regio impurity: 0.57%, Bromo impurity: 0.28%
Example 8: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine dihydrochloride (8.4 gm) were added to the reaction mixture at 26°C. The reaction mixture was heated to '\ 00 °C and maintained at that temperature for 4 hours. The reaction mixture was cooled to 30 °C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 200 mL of 10% aqueous acetic acid solution and stirred for 30 minutes at 28 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) were added drop- wise at 28°C (pH is > 10). The mixture was stirred for one hour at 28°C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and divided into 5 equal parts.
Part 1 : The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (16 mL) was added to the residue stirred for 30 minutes at 28 °C and 48 mL of MTBE was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 8 mL of MTBE and dried the compound at 65 °C under vacuum to give 3.0 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.46%, Regio impurity: 0.37%, Bromo impurity: 0.03%
Part 2: The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (24 mL) was added to the residue stirred for 30 minutes at 28 °C and the resulted solution was cooled to 5°C and stirred for 1 hour. Filtered the compound and washed with 5 mL of chilled methanol and dried the compound at 65°C under vacuum to give 3.0 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.41 %, Regio impurity: 0.38%, Bromo impurity: 0.03%
Part 3: The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (20 mL) was added to the residue stirred for 30 minutes at 28 °C and 20 mL of MTBE was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 8 mL of MTBE and dried the compound at 65 °C under vacuum to give 2.8 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.47%, Regio impurity: 0.36%, Bromo impurity: 0.03%.
Part 4: The organic layer was distilled off completely under vacuum at 45 °C. Isopropyl alcohol (8 mL) was added to the residue and distilled off completely under vacuum at 45 °C. Methanol (16 mL) was added to the residue stirred for 30 minutes at 28 °C and 16 mL of isopropyl alcohol was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 4 mL of isopropyl alcohol and dried the compound at 65 °C under vacuum to give 2.9 gm of Linagliptin. PXRD pattern: Fig. 1 .
Chemical Purity: 99.44%, Regio impurity: 0.38%, Bromo impurity: 0.02%.
Part 5: The organic layer was distilled off completely under vacuum at 45 °C. Ethyl acetate (8 mL) was added to the residue and distilled off completely under vacuum at 45 °C. Ethyl acetate (16 mL) was added to the residue stirred for 30 minutes at 28°C and 16 mL of methanol was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 4 mL of ethyl acetate and dried the compound at 65 °C under vacuum to give 0.7 gm of Linagliptin. PXRD pattern: Fig. 2.
Chemical Purity: 99.57%, Regio impurity: 0.29%, Bromo impurity: 0.02%
Example 9: Purification of Linagliptin
Linagliptin (3.5 gm) was dissolved in 10% aqueous acetic acid and stirred for 15 minutes. Dichloromethane (50 mL) was added to the solution and stirred for 30 minutes. The aqueous layer was separated and the pH of this layer was adjusted to 8.5 using 10% aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (50 mLx2). The dichloromethane was evaporated under vacuum to give 3 gm of Linagliptin.
Example 10: Purification of Linagliptin
Linagliptin (31 gm) and methanol (124 mL) were charged into 500 mL round bottomed flask and the solution was heated to 40 °C and stirred for 60 minutes. Charcoal (3 gm) was added to the clear solution and stirred for 30 minutes. The solution was filtered through Hy-flow and the Hy-flow bed was washed with methanol (30 mL). Filtrate was charged into 1000 mL round bottomed flask and methyl tertiary butyl ether was added drop-wise to the solution and stirred for 2 hours at 30 °C. The precipitate so formed was filtered and the wet cake was washed with methyl tertiary butyl ether (30 mL) to get 25.6 gm of pure Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.57%, Chiral purity: 99.73%, Regio impurity: 0.10%, Bromo impurity: 0.1 %
Example 1 1 : Purification of Linagliptin
Linagliptin (4 gm) and methanol (24 mL) were charged into 100 mL round bottomed flask and the solution is heated to 50 °C and stirred for 60 minutes. Methyl tertiary butyl ether (MTBE, 80mL) was charged into 500 mL round bottomed flask and the methanol solution containing linagliptin was added drop-wise at 27 °C and stirred for 2 hours at same temperature. The precipitate formed was filtered and the wet cake was washed with methyl tertiary butyl ether (8 mL) to get 2.6 gm of pure Linagliptin. PXRD pattern: Fig. 2, Bromo impurity content: 0.04%.
Example 12: Purification of Linagliptin
a) Preparation of linagliptin-(D)-tartrate
Linagliptin (10 gm) and methanol (300 mL) were charged into 1000 mL round bottomed flask and (D)-tartaric acid solution (3.3 gm of (D)-tartaric acid in 100 mL of methanol) was added at 26 °C. The solution was heated to 65 °C and stirred for 60 minutes. The solution was cooled to 28 °C and stirred for 2 hours at 27 °C. The precipitate formed was filtered and the wet cake was washed with methanol (20 mL) and the solid was dried under vacuum at 55°C to get 8.3 gm of Linagliptin-(D)-tartrate. PXRD pattern: Fig. 4. Chemical Purity: 99.72%, Chiral purity: 99.89%, Regio impurity: 0.08%, Bromo impurity: 0.05%, S-isomer: 0.1 1%.
b) Isolation of pure Linagliptin
Linagliptin-(D)-tartrate (8 gm) and water (100 mL) were charged into 1000 mL round bottomed flask and stirred for 30 minutes at 26 °C. Dichloromethane (80 mL) was added to the solution and cooled to 5°C. Aqueous sodium hydroxide solution (0.6 gm of NaOH is added to 20 mL of water) was added to the mixture at 5°C and maintained for 1 hour. Layers were separated and aqueous layer was extracted with dichloromethane (20 mL). Combined both organic layers and dried over sodium sulphate and distilled off the organic layer under vacuum at 45 °C. Hexane (20 mL) was added to the crude and stirred for 1 hour at 26°C. The precipitate was filtered and washed with 4 mL of hexane and dried the compound at 60°C under vacuum to give 6 gm of pure Linagliptin. PXRD pattern: Fig. 2, Chemical Purity: 99.67%, Chiral purity: 99.85%, (S)-isomer content: 0.1 5%, Regio impurity: 0.09%, Bromo impurity: 0.07%.
PATENT
http://www.google.com/patents/US20130123282
- [0181]8-Bromo-3-methylxanthine was reacted with 1-bromo-2-butyne in the presence of base in a mixture of N-methyl pyrrolidone and toluene mixture. The reaction mixture was heated overnight. The reaction completion was determined, and the mixture was then cooled to ambient temperature. A solid precipitate formed on cooling precipitation. The product, 3-Methyl-7-(2-butyne-1-yl)-8-bromoxanthine, having greater than 95% purity was isolated by filtration and washed with toluene.
- Example
34Preparation of
(R)-8-(3-amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione
(Form-XXII): A. 3-Methyl-7-(2-butyne-1-yl)-8-bromoxanthine
- [0182]3-Methyl-7-(2-butine-1-yl)-8-bromoxanthine was reacted with 2-(chloromethyl)-4-methylquinazoline in the presence of base under phase transfer catalyst using a N-methyl pyrrolidone/toluene mixture as the reaction solvent. The reaction mixture was heated overnight. When the reaction was complete, the reaction mixture was cooled to ambient temperature. A solid precipitate formed and was separated by filtration and washed with toluene and then with water to provide the product, 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione having more than 97% purity.
- [0183](R)-3-N-tert-Butoxycarbonylaminopiperidine was reacted with 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione in the presence of base. The reaction mixture was heated overnight. When the reaction was complete, the reaction mixture was cooled to ambient temperature. The cooled reaction mixture was washed several times with water and separated. The resulting 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine organic solution was greater than 95%. Purified by HPLC. An excess of aqueous HCl solution was added to the obtained 1-[(4-methylquinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine organic solution. The resulting mixture was stirred under heating until complete conversion was observed. Aqueous base was added to the reaction. The resulting mixture was stirred and separated. The organic phase was washed with aqueous base and separated. A non-polar or moderately polar solvent was added to the resulting organic phase. The mixture was partially concentrated to achieve precipitation, and the concentrated mixture was cooled and filtered to provide the wet crude product. The crude product was re-crystallized from alcohol, filtered and dried in vacuum oven with heating to afford dry solid Form-XXII of (R)-8-(3-amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione having more than 98% purity.
Clinical trials
Results in 2010 from a Phase III clinical trial of linagliptin showed that the drug can effectively reduce blood sugar.[2]
Scheme:
. J. Med Chem 2009, 52, 6433..
J. Med Chem 2007, 50, 6450...

References
- H. Spreitzer (September 1, 2008). "Neue Wirkstoffe - BI-1356". Österreichische Apothekerzeitung (in German) (18/2008): 918.
- Wang, Y, Serradell, N, Rosa, E, Castaner, R (2008). "BI-1356". Drugs of the Future 33 (6): 473–477. doi:10.1358/dof.2008.033.06.1215244.
- ^ "FDA Approves Type 2 Diabetes Drug from Boehringer Ingelheim and Lilly". 3 May 2011.
- "Four Phase III Trials Confirm Benefits of BI’s Oral, Once-Daily Type 2 Diabetes Therapy". Genetic Engineering & Biotechnology News. 28 June 2010.
| CN101735218A * | Dec 17, 2009 | Jun 16, 2010 | 廖国超 | Piperidine carbamic acid ester derivative and application thereof |
| US7407955 | Aug 12, 2003 | Aug 5, 2008 | Boehringer Ingelheim Pharma Gmbh & Co., Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US20040097510 * | Aug 12, 2003 | May 20, 2004 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US20090192314 | Mar 30, 2009 | Jul 30, 2009 | Boehringer Ingelheim International Gmbh | Process for the preparation of chiral 8-(3-aminopiperidin-1yl)-xanthines |
| WO2005085246A1 * | Feb 12, 2005 | Sep 15, 2005 | Boehringer Ingelheim Int | 8-[3-amino-piperidin-1-yl]-xanthine, the production thereof and the use in the form of a dpp inhibitor |
| Reference | ||
|---|---|---|
| 1 | CHIRALITY vol. 7, 1995, pages 90 - 95 | |
| 2 | * | JEAN L ET AL: "A convenient route to 1-benzyl 3-aminopyrrolidine and 3-aminopiperidine", TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 42, no. 33, 13 August 2001 (2001-08-13), pages 5645-5649, XP004295831, ISSN: 0040-4039, DOI: DOI:10.1016/S0040-4039(01)00985-6 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014033746A2 * | Aug 6, 2013 | Mar 6, 2014 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Process for the preparation of dipeptidylpeptidase inhibitors |
| WO2014059938A1 * | Oct 17, 2013 | Apr 24, 2014 | 2Y-Chem, Ltd. | Method for preparing important intermediate of linagliptin |
| WO2014097314A1 * | Dec 16, 2013 | Jun 26, 2014 | Mylan Laboratories Ltd | An improved process for the preparation of linagliptin |
| WO2010072776A1 * | Dec 22, 2009 | Jul 1, 2010 | Boehringer Ingelheim International Gmbh | Salt forms of organic compound |
| CN101784270A * | Aug 15, 2008 | Jul 21, 2010 | 贝林格尔.英格海姆国际有限公司 | Pharmaceutical composition comprising a glucopyranosyl-substituted benzene derivative |
| CN102127080A * | Nov 2, 2005 | Jul 20, 2011 | 贝林格尔.英格海姆国际有限公司 | Method for producing chiral 8-(3-amino-piperidin-1-yl)-xanthines |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015067539A1 * | Oct 31, 2014 | May 14, 2015 | Chemelectiva S.R.L. | Process and intermediates for the preparation of linagliptin |
| WO2015087240A1 | Dec 9, 2014 | Jun 18, 2015 | Ranbaxy Laboratories Limited | Process for the preparation of linagliptin and an intermediate thereof |
| WO2015107533A1 * | Sep 1, 2014 | Jul 23, 2015 | Harman Finochem Limited | A process for preparation of 1h-purine-2,6-dione, 8-[(3r)-3-amino-1-piperidinyl]-7 (2-butyn-1-yl)-3,7-dihydro-3-methyl-1-[(4-methyl-2quinazolinyl) methyl] and its pharmaceutically acceptable salts |
| Eckhardt M, et al. 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J Med Chem. 2007; 50(26):6450-3. Pubmed ID: 18052023 | |
| 2. | Thomas L, et al. (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J Pharmacol Exp Ther. 2008; 325(1):175-82. Pubmed ID: 18223196 |
//////////BI-1356, BI1356, Linagliptin, Tradjenta, Trajenta, DPP-IV, DPP-4 inhibitor
No comments:
Post a Comment