

TGR 1202, TGR-1202-101, RP 5264
AK173784;
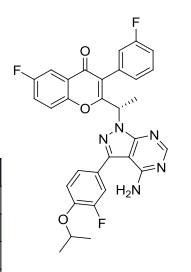
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one,
2-[(1S)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one
CAS 1532533-67-7, 1514919-95-9
| Molecular Formula: | C31H24F3N5O3 |
|---|---|
| Molecular Weight: | 571.54917 g/mol |
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta
inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available,
PI3K delta inhibitor, targeting the delta isoform with nanomolar potency
and several fold selectivity over the alpha, beta, and gamma isoforms
of PI3K.
TG Therapeutics, under license from Rhizen
Pharmaceuticals, is developing TGR-1202 (structure shown; formerly
RP-5264), a lead from a program of PI3K delta inhibitors, for the
potential oral treatment of hematological cancers including Hodgkin
lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia
(CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
| Rhizen Pharmaceuticals S.A. | |
| Description | Phosphoinositide 3-kinase (PI3K) delta inhibitor |
CLINICAL TRIALS……….https://clinicaltrials.gov/search/intervention=TGR-1202
B-cell lymphoma; Chronic lymphocytic leukemia;
Hematological neoplasm; Hodgkins disease; Mantle cell lymphoma;
Non-Hodgkin lymphoma
Phosphoinositide-3 kinase delta inhibitor
SYNTHESIS


Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
| Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) —
Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement
for TGR-1202, a novel next generation PI3K-delta inhibitor. TG
Therapeutics exercised its option for early conversion to a licensing
agreement from a 50:50 joint venture partnership.In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen
stated, “We are extremely happy and take pride in discovering a novel,
next generation, once-daily PI3K-delta inhibitor under active
development led by TG Therapeutics. We are encouraged by the progress
of TRG-1202 to date, and the speed at which TG Therapeutics is
developing the asset in various hematological malignancies. We look
forward to the day this novel drug reaches cancer patients in need of
new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative,
clinical-stage biopharmaceutical company focused on the discovery and
development of novel therapeutics for the treatment of cancer, immune
and metabolic disorders. Since its establishment in 2008, Rhizen has
created a diverse pipeline of proprietary drug candidates targeting
several cancers and immune associated cellular pathways. Rhizen is
headquartered in La-Chaux-de-Fonds, Switzerland. For additional
information, please visit Rhizen’s website, www.rhizen.com.

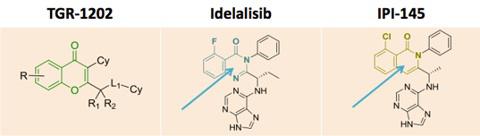
TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
IPI 145
PATENTS
WO 2011055215http://www.google.com/patents/WO2011055215A2?cl=en

WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:

Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)

7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)

1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1

The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8

PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,

Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one

Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate


Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene

chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
http://www.google.com/patents/WO2014006572A1?cl=en
 B1 IS DESIRED
B1 IS DESIRED(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
-
To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
-
Various other acid addition salts of compound B1 were prepared as provided in Table 1.
-
TABLE 1 Melting Point Acid Method of preparation (° C.) Hydro- Compound B1 (1 eq.) dissolved in THF, 130-132 chloric excess HCl/Et2O was added, the clear acid solution obtained was evaporated completely. The residue obtained was washed with water. p- Compound B1 (1 eq.) dissolved in 138-141° C. Toluene- isopropyl alcohol (IPA), refluxed for sulfonic 30 min., acid (1.1 eq.) in IPA was added, acid the clear solution obtained was evaporated completely. The residue obtained was washed with water. Benzene- Compound B1 (1 eq.) dissolved in IPA, 170-172 sulphonic refluxed for 30 min., acid(1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Maleic Compound B1 (1 eq.) dissolved in IPA, 107-109 acid refluxed for 30 min., acid (1.1 eq.) in IPA was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Camphor Compound B1 (1 eq.) dissolved in IPA, 120-121 sulfonic refluxed for 30 min., acid (1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Sulphuric Compound B1 (1 eq.) dissolved in IPA, 125-127 acid refluxed for 30 min., acid(1.1 eq.) in IPA was added, the clear solution obtained was evaporated completely. The residue obtained was washed with water.
REFERENCES
WO 2014/006572 and U.S. Patent Publication No. 2014/0011819,
http://www.tgtherapeutics.com/O’ConnorTGR202Single%20AgentEHA&Lugano2015.pdf
-
TGR-1202: Phase I/II started 09/28/2015
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Phase I/II started 09/28/2015
Week in Review, Clinical StatusLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 … -
The Daily Extra, Company NewsTG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
-
Targets & Mechanisms: The battle for IRAK 04/23/2015
Nimbus, Aurigene and TG Therapeutics are chasing IRAK4 inhibitors for cancerBC Innovations, Targets & MechanismsNow that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year. -
TGR-1202: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer … -
TGR-1202: Phase I started 12/15/2014
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Rhizen, TG Therapeutics deal 12/08/2014
Week in Review, DealsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
| Patent | Submitted | Granted |
|---|---|---|
| NOVEL SELECTIVE PI3K DELTA INHIBITORS [US2014011819] | 2013-07-02 | 2014-01-09 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
OSI-420 (CP-473420) is an active metabolite of erlotinib which is an orally active EGFR tyrosin kinase inhibitor with IC50 of 2 and 20 nM for the inhibition of human EGFR and EGFR autophosphorylation in tumor cells. OSI-420
ReplyDelete