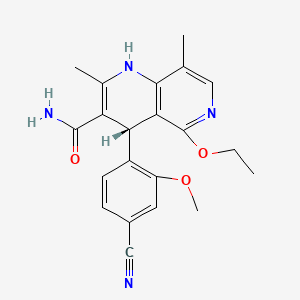
Finerenone
Finerenone; UNII-DE2O63YV8R; BAY 94-8862; DE2O63YV8R; 1050477-31-0
| C21H22N4O3 | |
| MW | 378.42438 g/mol |
|---|
(4s)-4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1-6-naphthyridine-3-carbox-amide
Bayer Corp
Mineralocorticoid receptor antagonist
phase III in January 2016, for treating diabetic kidney disease and chronic heart failure in patients with worsening chronic cardiac insufficiency
Used as mineralocorticoid receptor antagonist for treating heart failure and diabetic nephropathy.
SYNTHESIS

Finerenone (INN, USAN) (developmental code name BAY-94-8862) is a non-steroidal antimineralocorticoid that is in phase IIIclinical trials for the treatment of chronic heart failure as of October 2015. It has less relative affinity to other steroid hormone receptors than currently available antimineralocorticoids such as eplerenone and spironolactone, which should result in fewer adverse effects like gynaecomastia, impotence, and low sex drive.[1][2]
Pharmacology
Finerenone blocks mineralocorticoid receptors, which makes it a potassium-sparing diuretic.
This table compares inhibitory (blocking) concentrations (IC50, unit: nM) of three antimineralocorticoids. Mineralocorticoid receptor inhibition is responsible for the desired action of the drugs, whereas inhibition of the other receptors potentially leads to side effects. Lower values mean stronger inhibition.[1]
| Spironolactone | Eplerenone | Finerenone | |
|---|---|---|---|
| Mineralocorticoid receptor | 24 | 990 | 18 |
| Glucocorticoid receptor | 2400 | 22,000 | >10,000 |
| Androgen receptor | 77 | 21,200 | >10,000 |
| Progesterone receptor | 740 | 31,200 | >10,000 |
The above-listed drugs have insignificant affinity for the estrogen receptor.
Chemistry
Unlike currently marketed antimineralocorticoids, finerenone is not a steroid but a dihydropyridine derivative.
Research
The drug is also being investigated in early trials for the treatment of diabetic nephropathy.[3]
PAPER
Discovery of BAY 94-8862: A Nonsteroidal Antagonist of the Mineralocorticoid Receptor for the Treatment of Cardiorenal Diseases
Article first published online: 12 JUL 2012
DOI: 10.1002/cmdc.201200081

ChemMedChem
Abstract
Aldosterone is a hormone that exerts manifold deleterious effects on the kidneys, blood vessels, and heart which can lead to pathophysiological consequences. Inhibition of the mineralocorticoid receptor (MR) is a proven therapeutic concept for the management of associated diseases. Use of the currently marketed MR antagonists spironolactone and eplerenone is restricted, however, due to a lack of selectivity in spironolactone and the lower potency and efficacy of eplerenone. Several pharmaceutical companies have implemented programs to identify drugs that overcome the known liabilities of steroidal MR antagonists. Herein we disclose an extended SAR exploration starting from cyano-1,4-dihydropyridines that were identified by high-throughput screening. Our efforts led to the identification of a dihydronaphthyridine, BAY 94-8862, which is a potent, selective, and orally available nonsteroidal MR antagonist currently under investigation in a clinical phase II trial.
PATENT
WO2008104306,
Lars Baerfacker, BELOW

Peter Kolkhof, BELOW

Karl-Heinz Schlemmer, Rolf Grosser, Adam Nitsche,Martina Klein, Klaus Muenter, Barbara Albrecht-Kuepper, Elke Hartmann,
EXAMPLES
Example 1
4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2-methyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxamide

100 mg (ca. 0:24 mmol) of the compound from Example 23A are initially charged in 3 ml DMF. Is 2.94 mg Then (0.024 mmol) of 4-N, N-dimethylaminopyridine and 340 ul of ammonia (28 wt .-% - solution in water, 2:41 mmol) and 3 h at 100 0 C temperature. After cooling, the crude product is purified directly by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 → 95: 5). There are 32 mg (37% d. Th.) The title connection receive.
LC-MS (Method 3): R, = 1:57 min; MS (EIPOS): m / z = 365 [M + H] +
1 H-NMR (300 MHz, DMSOd6): δ = 1:07 (t, 3H), 2.13 (s, 3H), 3.83 (s, 3H), 4:04 (m, 2H), 5:36 (s, IH), 6:42 (d, IH), 6.66 (br. s, 2H), 7.18 (d, IH), 7.29 (dd, IH), 7:38 (d, IH), 7.67 (d, IH), 8.80 (s, IH).
Example 2
4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,7-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxamide

640 mg (1.69 mmol) of the compound from Example 27A are initially charged in 30 ml of ethyl acetate, 342 mg (2.11 mmol) l, r-carbonyldiimidazole and then stirred overnight at room temperature. A TLC check (silica gel; mobile phase: cyclohexane / ethyl acetate 1: 1 or dichloromethane / methanol 9: 1) shows complete conversion. The volatile components are removed on a rotary evaporator and the residue taken up in 20 ml DMF. Subsequently, 2.36 ml of ammonia (28 wt .-% - solution in water, 16.87 mmol) was added and the reaction mixture for 8 hours at 50 0 C temperature. The solvent is distilled off under reduced pressure and the residue purified by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 -> 95: 5). This gives 368 mg (58% d. Th.) Of the title compound.
LC-MS (method 7): R t = 1.91 min; MS (EIPOS): m / z = 379 [M + H] +
1 H-NMR (300 MHz, DMSO-d 6): δ = 1:05 (t, 3H), 2.13 (s, 3H), 2.19 (s, 3H), 3.84 (s, 3H), 4:02 (q, 2H) , 5:32 (s, IH), 6.25 (s, IH), 6.62 (br. s, 2H), 7.16 (d, IH), 7.28 (dd, IH), 7:37 (d, IH), 8.71 (s, IH ).
Example 3
e 'f 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,7-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox- amide [(-) - enantiomer and (+) - enantiomer \

The racemate of Example 2 can be separated on a preparative scale by chiral HPLC into its enantiomers [column: Chiralpak IA, 250 mm x 20 mm; Eluent: methyl tert-butyl ether / methanol 85: 15 (v / v); Flow: 15 ml / min; Temperature: 30 0 C; UV detection: 220 Dm].
(-) - Enantiomer:
HPLC: R, = 5.28 min, ee> 98% [column: Chiralpak IA, 250 mm x 4.6 mm; Eluent: methyl tert-butyl ether / methanol 80:20 (v / v); Flow: 1 ml / min; Temperature: 25 0 C; UV detection: 220 nm];
specific optical rotation (chloroform, nm 589, 19.8 ° C, c = 0.50500 g / 100 ml): -239.3 °.
A single crystal X-ray structural analysis revealed a ^ -configuration at C * for this enantiomer - atom.
(+) - Enantiomer:
HPLC: R = 4:50 min, ee> 99% [column: Chiralpak IA, 250 mm x 4.6 mm; Eluent: methyl tert-butyl ether / methanol 80:20 (v / v); Flow: 1 ml / min; Temperature: 25 ° C; UV detection: 220 nm];
specific optical rotation (chloroform, nm 589, 20 0 C, c = 0.51000 g / 100 ml): + 222.7 °.
Example 4
4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxamide

1:46 g (3.84 mmol) of the compound from Example 3oA are introduced into 50 ml of ethyl acetate, 777 mg (4.79 mmol) l, r-carbonyldiimidazole and then stirred overnight at room temperature. A TLC check (silica gel; eluent: ethyl acetate) shows complete conversion. The volatile components are removed on a rotary evaporator and the residue taken up in 20 ml DMF.Then 10.74 ml of ammonia (28 wt% solution in water, 76.8 mmol) was added and the reaction mixture heated for 30 minutes at 100 0 C. The solvent is distilled off under reduced pressure and the residue purified by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 -> 95: 5). After concentrating the product fractions, the residue in 40 ml of dichloromethane / methanol (1: 1 v / v) and treated with 100 ml of ethyl acetate. The solvent is concentrated to a volume of about 20 ml, whereupon the product crystallized. The precipitate is filtered off and washed with a little diethyl ether.After drying at 40 0 C in a vacuum oven obtained 1:40 g (96%. Th.) The title connection.
LC-MS (Method 3): R, = 1.64 min; MS (EIPOS): m / z = 379 [M + H] +
1 H-NMR (300 MHz, DMSOd6): δ = 1:05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H) , 5:37 (s, IH), 6.60-6.84 (m, 2H), 7.14 (d, IH), 7.28 (dd, IH), 7:37 (d, IH), 7:55 (s, IH), 7.69 (s, IH ).
Example 5
e "M- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox- amide [(-) - enantiomer and (+ ) enantiomer]

The racemate of Example 4 can be separated on a preparative scale by chiral HPLC into its enantiomers [column: 680 mm x 40 mm; Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 80 ml / min; UV detection: 260 nm].
(-) - Enantiomer:
HPLC: R = 2:48 min, ee = 99.6% [column: 250 mm x 4.6 mm; Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 2 ml / min; UV detection: 260 nm];
specific optical rotation (chloroform, nm 589, 19.7 ° C, c = 0.38600 g / 100 ml): -148.8 °.
A single crystal X-ray structure analysis showed this enantiomer S configuration at C * - atom.
(+) - Enantiomer:
HPLC: R = 4:04 min, ee = 99.3% [column: 250 mm x 4.6 mm; Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 2 ml / min; UV detection: 260 nm];
specific optical rotation (chloroform, nm 589, 19.8 ° C, c = 0.36300 g / 100 ml): + 153.0 °.
PATENT
The present invention relates to a novel and improved process for preparing 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3-carbox- amide of formula (I)

as well as the preparation and use of crystalline modification I of (4S) - 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3- carbox-amide of formula (I).
The compound of formula (I) acts as a non-steroidal mineralocorticoid receptor antagonist and can be used as agents for the prophylaxis and / or treatment of cardiovascular and renal diseases such as heart failure and diabetic nephropathy.
The compound of formula (I) and their preparation process are described in WO 2008/104306 and ChemMedChem 2012 7, described in 1385, in both publications a detailed discussion of research synthesis is disclosed. A disadvantage of the synthesis described there is the fact that this synthesis is not suitable for another large-scale process, since many steps in very high dilution, at very high reagent surpluses and thus run on a relatively low overall yield. Furthermore, many chromatographic cleanings are necessary, which are usually very expensive and require a high consumption of solvents, are costly and which should therefore be avoided if possible.Some stages can not be realized due to safety and procedural difficulties.
There is therefore a need for an industrially viable synthesis, reproducible in high overall yield, low production costs and high purity provides the compound of formula (I) and complies with all regulatory requirements in order to supply the clinical trials on drug and for subsequent regulatory submission to be used.
With the present invention a very efficient synthesis has been found, which allows to meet the above requirements.
In the publication ChemMedChem 2012 7, in which the research synthesis of the compound of formula (I) disclosed in 1385, the compound of formula (I), starting from vanillin prepared in 10 steps with an overall yield of 3.76% of theory , The compound of formula (I) was obtained by evaporation of the chromatography fractions as an amorphous solid, a defined process Kristalhsations- the stage for polymorphism-setting has not been described.
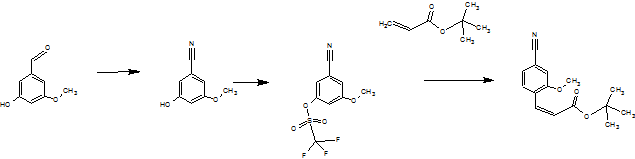
The following Scheme 1 shows the known process for preparing the compound of formula (I).

(II) (HI) (IV)

(V) (VI)



(XIII) (I)
Scheme 1: synthesis research of the compound of formula (I)
There are used 3 chromatographic purifications, and a chiral chromatography step to separate the enantiomers of the racemate of formula (XIII). The steps run partially in very high dilution and using very large amounts of reagent.
Thus, in particular the sequence of the preparation of the nitrile aldehyde intermediate (VI), which occupies a central role in the synthesis of atom not economically acceptable.
Furthermore, not to apply this process to an industrial scale, since [=> (IV) (III)] and excesses of acrylic acid tert-butyl ester are used for a very expensive reagents such as trifluoromethanesulfonic anhydride. When upscaling the Heck reaction (IV) => (V) formed in the boiler, a plastic similar residue resulting from the polymerization of acrylic acid tert.butyl ester used in excess. This is not acceptable in the technical implementation, there is a risk that there may be a Rührerbruch and it would lead to strong to remove residues in the agitators.
The subsequent cleavage of the double bond with sodium and the highly toxic osmium tetroxide is to be avoided since there is a delay of reaction and thereby caused to a strongly exothermic and connected with that comes a runaway under the test conditions described.
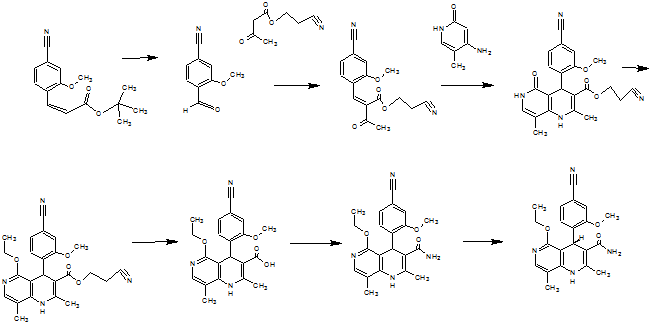
Scheme 2 illustrates the new process of the invention that the compound of formula (I) in 9 levels in 27.7% d. Th. Total yield without a chromatographic
Purification of intermediates supplies.




Scheme 2: According to the Invention for preparing the compound of formula (I).
Examples
example 1
Methyl 4-bromo-2-methoxybenzoate (XV)
3.06 kg (22.12 mol) potassium carbonate are placed in 1 acetone 3.6 and heated to reflux. To this suspension is metered in 1.2 kg of 4-bromo-2-hydroxybenzoic acid (5.53 mol) suspended in 7.8 1 of acetone and rinsed with 0.6 1 acetone. The mixture is heated for one hour under reflux (vigorous evolution of gas!). is boiled for 2.65 kg (21.01 mol) Dimethylsufat over 4 hours then metered. 2.5 hours then is stirred under reflux. The solvent is distilled off to a large extent (up to the stirrability) and returns to 12 1 toluene, then the remaining acetone is distilled off at 110 ° C. There are about 3 1 distillate distilled, these are supplemented by the addition of a further 3 1 toluene to approach. Allow to cool to 20 ° C and are 10.8 1 water were added and agitated vigorously. The organic phase is separated and the aqueous phase extracted again with 6.1 1 of toluene. The combined organic phases are washed with 3 1 of saturated sodium chloride solution, and the toluene phase is concentrated to about 4 first A quantitative analysis by evaporating a subset results converted a yield 1.306 kg (96.4% of theory). The solution is used directly in the next stage.
HPLC method A: RT about 11.9 min.
MS (EIPOS): m / z = 245 [M + H] +
H NMR (400 MHz, CD 2 C1 2 ): δ = 3.84 (s, 3H), 3.90 (s, 3H), 7:12 to 7:20 (m, 2H), 7.62 (d, 1H).
example 2
4-bromo-2-methoxybenzaldehyde (XVI)
It puts 1.936 kg (6.22 mol) 65% Red- Al solution in toluene with 1.25 1 of toluene at -5 ° C before. To this solution was dosed 0.66 kg (6.59 mol) of 1-methylpiperazine and rinsed with 150 ml of toluene, the temperature keeps you here from -7 to -5 ° C.. It is allowed for 30 minutes at 0 ° C. for. This solution is then dosed to a solution of 1.261 kg (5.147 mol) of methyl 4-bromo-2-methoxybenzoate (XV), dissolved in 4 1 of toluene, the temperature is maintained at - 8-0 ° C. Rinse twice with 0.7 1 of toluene and stirred for 1.5 hours at 0 ° C to. For working up, dosed to a 0 ° C cold aqueous sulfuric acid (12.5 1 water + 1.4 kg of conc. Sulfuric acid). The temperature should rise to a maximum of 10 ° C (slow dosage). The pH is, if necessary, by addition of further sulfuric acid to a pH of the first The organic phase is separated and extracted the aqueous phase with 7.6 1 of toluene. The combined organic phases are washed with 5.1 1 of water and then substantially concentrated and the residue taken up with 10 1 DMF. The mixture is concentrated again to about 5 1 volume. A quantitative analysis by evaporating a subset results converted a yield 1.041 kg (94.1% of theory). The solution is used directly in the next stage.
HPLC method A: RT approximately 12.1 min.
MS (EIPOS): m / z = 162 [M + H] +
X H-NMR (CDCl, 400MHz): δ = 3.93 (3H, s), 7.17 (2H, m), 7.68 (1H, d), 10:40 (1H, s)
example 3
4-formyl-3-methoxybenzonitrile (VI)
719 g (3.34 mol) of 4-bromo-2-methoxybenzaldehyde (XVI) as a solution in 4.5 1 of DMF with 313 g (0.74 mol) of potassium hexacyanoferrate (K4 [Fe (CN) 6]) and 354 g submitted (3.34 mol) of sodium carbonate and a further 1.2 1 of DMF and 3.8 g (0.017 mol) of palladium acetate. It is stirred for 3 hours at 120 ° C. Allow to cool to 20 ° C and are 5.7 1 water to approach. It is extracted with 17 1 ethyl acetate, and the aqueous phase is washed again with 17 1 of ethyl acetate to. The organic phases are combined and substantially concentrated with 5 1 of isopropanol was added and concentrated to about 2 1st The mixture is heated to boiling and dripping 2 1 of water.Allow to cool to 50 ° C and are again added 2 1 water. It is cooled to 3 ° C and stirred for one hour at this temperature. The product is filtered and washed with water (2 times 1.2 1) washed. It is dried at 40 ° C under vacuum.
Yield: 469 g (87% of theory.) Of a beige solid.
HPLC method A: RT about 8.3 min.
MS (EIPOS): m / z = 162 [M + H] +
1H-NMR (300 MHz, DMSO-d6): δ = 3.98 (s, 3H), 7:53 (d, 1H), 7.80 (s, 1H), 7.81 (d, 1H), 10:37 (s, 1H).
example 4
2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -2,8-dimethyl-5-oxo-l, 4,5,6-tett ^
din-3-carboxylate (X)
option A
1.035 kg (6.422 mol) of 4-formyl-3-methoxybenzonitrile (VI), 1.246 kg (8.028 mol) of 2-Cyanefhyl 3-oxobutanoate, 54.6 g (0.642 mol) of piperidine and 38.5 g (0.642 mol) of glacial acetic acid are heated under reflux on a water in 10 1 dichloromethane 6.5 hours. Allow to cool to room temperature and the organic phase was washed 2 times with 5 1 water. Subsequently, the dichloromethane phase is concentrated under atmospheric pressure and the still stirrable residue with 15.47 kg of 2-butanol and 0.717 kg (5.78 mol) of 4-amino-5-methylpyridone added. The residual dichloromethane is distilled off until an internal temperature of 98 ° C is reached. Then, 20 hours, heated under reflux. It is cooled to 0 ° C, can be 4 hours at this temperature is stirred and filtered off the product. It is dried at 40 ° C under vacuum to the carrier gas.
Yield: 2.049 kg (87.6% of theory based on 4-amino-5-methylpyridone, since this component is used in deficiency) of a slightly yellowish colored solid.
HPLC method A: RT about 9.7 min.
MS (EIPOS): m / z = 405 [M + H] +
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 2:03 (s, 3H), 2:35 (s, 3H), 2.80 (m, 2H), 3.74 (s, 3H), 4:04 (m, 1H), 4.11 (m, 1H), 5.20 (s, 1H), 6.95 (s, 1H), 7.23 (dd, 1H), 7:28 to 7:33 (m, 2H), 8.18 (s, 1H), 10.76 (s, 1H) ,
variant B
1.344 kg (8.34 mol) of 4-formyl-3-methoxy-benzonitrile (VI), 71 g (0.834 mol) piperidine and 50.1 g (0.834 mol) of glacial acetic acid are introduced into 6 1 of isopropanol at 30 ° C within 3 hours, a solution of 1.747 kg (11.26 mol) of 2-cyanoethyl 3-oxobutanoate metered in 670 ml of isopropanol. Stirring an hour after at 30 ° C. It is cooled to 0-3 ° C and stirred at 0.5 hours. the product is filtered off and washed 2 times with 450 ml of cold isopropanol to. For yield determination is under vacuum at 50 ° C. (2.413 kg, 97% of theory..); but it is usually due to the high yield continued to work directly with the isopropanol-moist product. For this, the product is taken up with 29 1 of isopropanol and 1.277 kg (7.92
mol) of 4-amino-5-methylpyridone added, followed by 24 internal temperature under about 1.4 bar overpressure in the closed vessel is heated at 100 ° C h. It is cooled by a ramp within 5 h at 0 ° C. stirred for 3 hours at 0 ° C. It is filtered off and washed with 2.1 1 of cold isopropanol. It is dried under vacuum at 60 ° C.
Yield: 2.819 kg (88% of theory based on 4-amino-5-methylpyridone, since this component is used in deficiency) of a slightly yellowish colored solid.
HPLC method A: RT about 9.7 min.
MS (EIPOS): m / z = 405 [M + H] +
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 2:03 (s, 3H), 2:35 (s, 3H), 2.80 (m, 2H), 3.74 (s, 3H), 4:04 (m, 1H), 4.11 (m, 1H), 5.20 (s, 1H), 6.95 (s, 1H), 7.23 (dd, 1H), 7:28 to 7:33 (m, 2H), 8.18 (s, 1H), 10.76 (s, 1H) ,
example 5
2- cyanoethyl-4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI)
2.142 kg (5.3 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -2,8-dimefhyl-5-oxo-l, 4,5,6-tetrahydro-l, 6-naphthyridin-3 carboxylate (X) and 4.70 kg (29 mol) of triethyl orthoacetate are dissolved in 12.15 1 of dimethylacetamide and 157.5 grams of concentrated sulfuric acid was added. The mixture is heated for 1.5 hours at 115 ° C and then cooled to 50 ° C. At 50 ° C are added dropwise to 30 minutes 12.15 1 water. After complete addition the Titelbelbindung (XI) is treated with 10 g seeded and further added dropwise to 12.15 1 of water over 30 minutes at 50 ° C. It is cooled to 0 ° C (ramp, 2 hours) and stirred for 2 hours at 0 ° C to. The product is filtered, washed 2 times each with 7.7 1 of water and dried in vacuo at 50 ° C.
Yield: 2114.2 g (92.2% of theory) of a slightly yellowish colored solid.
HPLC Method B: RT 10,2 min.
MS (EIPOS): m / z = 433 [M + H] +
X H-NMR (300 MHz, DMSO-d 6 ): δ = 1.11 (t, 3H), 2.16 (s, 3H), 2:42 (s, 3H), 2.78 (m, 2H), 3.77 (s, 3H) , 4:01 to 4:13 (m, 4H), 5:37 (s, 1H), 7.25 (d, 1H), 7:28 to 7:33 (m, 2H), 7.60 (s, 1H), 8:35 (s, 1H).
Alternatively, the reaction in NMP (l-methyl-2-pyrrolidone) may be carried out
2- cyanoethyl-4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI)
2.142 kg (5.3 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -2,8-dimethyl-5-oxo-l, 4,5,6-tetrahydro-l, 6-naphthyridin-3 carboxylate (X) and 2.35 kg (14.5 mol) of triethyl orthoacetate are in 3.21 kg NMP (l-methyl-2-pyrrolidone) and dissolved 157.5 g of concentrated sulfuric acid was added. The mixture is heated for 1.5 hours at 115 ° C and then cooled to 50 ° C. At 50 ° C are added dropwise to 30 minutes 2.2 1 water. After complete addition the Titelbelbindung (XI) is treated with 10 g seeded and dropped further 4.4 1 of water over 30 minutes at 50 ° C. It is cooled to 0 ° C (ramp, 2 hours) and stirred for 2 hours at 0 ° C to. The product is filtered off, washed 2 times each with 4 1 of water and dried under vacuum at 50 ° C.
Yield: 2180.7 g (95.1% of theory) of a slightly yellowish colored solid.
HPLC Method B: RT 10,2 min.
example 6
4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3-carboxylic acid IXM
2.00 kg (4.624 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI ) are dissolved in a mixture of 12 1 THF and 6 1 of water and cooled to 0 ° C. To this solution, a sodium hydroxide solution is added in drops within 15 minutes at 0 ° C (prepared from 0.82 kg 45% aqueous. NaOH (9.248 mol) and 4.23 1 of water and stirred for 1.5 hours at 0 ° C to . The mixture is extracted 2 times with each 4.8 1 methyl tert-butyl and once with 4.8 1 of ethyl acetate. The aqueous solution is at 0 ° C with dilute hydrochloric acid (prepared from 0.371 kg 37% HCl and 1.51 1 water ) adjusted to pH 7. the mixture is allowed to warm to 20 ° C and adding an aqueous solution of 2.05 kg of ammonium chloride in 5.54 1 water. the mixture is stirred 1 hour at 20 ° C, the product filtered and 2 times with each each 1.5 1 water and washed once with 4 1 acetonitrile. It is dried at 40 ° C under vacuum to the carrier gas.
Yield: 1736.9 g (99% of theory..) Of an almost colorless powder (very slight yellow tinge).
HPLC Method C: RT: about 6.8 min.
MS (EIPOS): m / z = 380 [M + H]
X H-NMR (300 MHz, DMSO-d 6 ): δ = 1.14 (t, 3H), 2.14 (s, 3H), 2:37 (s, 3H), 3.73 (s, 3H), 4:04 (m, 2H) , 5:33 (s, 1H), 7.26 (m, 2H), 7:32 (s, 1H), 7:57 (s, 1H), 8.16 (s, 1H), 11:43 (br. s, 1H).
Alternative workup with toluene for extraction:
4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylic-isäure (XII)
2.00 kg (4.624 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI ) are dissolved in a mixture of 12 1 THF and 6 1 of water and cooled to 0 ° C. To this solution, a sodium hydroxide solution is added in drops within 15 minutes at 0 ° C (prepared from 0.82 kg 45% aqueous. NaOH (9.248 mol) and 4.23 1 of water and stirred for 1.5 hours at 0 ° C to . Add 5 L of toluene and 381.3 g Natiumacetat added and stirred vigorously. Allow to settle the phases and the organic phase is separated. the aqueous phase is adjusted with 10% hydrochloric acid to pH 6.9 (at about pH 9.5 is inoculated with 10 g of the title compound of). After completion of the precipitation of the product for one hour at 0 ° C is stirred and then filtered and washed twice with 4 1 of water and twice with 153 ml of toluene. the mixture is dried at 40 ° C under vacuum to carrier gas (nitrogen, 200 mbar. yield:.. 1719.5 g (98% of theory) of an almost colorless powder (very slight yellow tinge).
HPLC Method C: RT: about 6.8 min).
example 7
4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3-carboxamide
1.60 kg (4.22 mol) of 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylic-isäure ( XII) and 958 g (5.91 mol) of 1,1-carbodiimidazole be presented in 8 1 of THF and at 20 ° C 51 g (0.417 mol) of DMAP was added. Stirring for one hour at 20 ° C (gas evolution!) And then heated 2.5 hours 50 ° C. are added to this solution 2.973 kg (18.42 mol) of hexamethyldisilazane and boil for 22 hours under reflux. Man admits further 1.8 1 THF and cooled to 5 ° C. A mixture is prepared from 1.17 1 of THF and 835 g of water is metered in over 3 hours, so that the temperature is between 5 and 20 ° C remains. Then boiled for one hour under reflux, then cooled via a ramp (3 hours) at 0 ° C. and stirred for one hour at this temperature. The product is filtered off and washed 2 times with 2.4 1 THF and twice with 3.2 1 water. It is dried under vacuum at 70 ° C under a carrier gas.
Yield: 1.501 kg (. 94% of theory) of an almost colorless powder (very slight yellow tinge).
HPLC Method B: RT about 6.7 min.
MS (EIPOS): m / z = 379 [M + H]
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 1:05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H ), 5:37 (s, 1H), 6.60-6.84 (m, 2H), 7.14 (d, 1H), 7.28 (dd, 1H), 7:37 (d, 1H), 7:55 (s, 1H), 7.69 (s, 1H).
example 8
(4S) - 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy
carbox-amide (I) as a solution in acetonitrile / Methariol 40:60
Enantiomeric separation on a SMB unit
As a feed solution a solution corresponding to a concentration is used consisting of 50 g racemic 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridin-3 -carbox-amide (XIII) dissolved in 1 liter of a mixture of methanol / acetonitrile 60:40.
There is a SMB unit on a stationary phase: 20 chromatographed μιη Chiralpak AS-V. The pressure is 30 bar, as the eluent a mixture of methanol / acetonitrile 60:40 is used.
9.00 kg of 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (XII) are dissolved in 180 1 a mixture dissolved consisting of methanol / acetonitrile 60:40 and chromatographed by SMB. After concentrating the product-containing fractions, 69.68 liters of a 6.2% solution (corresponding to 4.32 kg (4S) - 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl- 1, 4-dihydro- 1, 6-naphthyridine-3-carbox-amide (I) as a solution in acetonitrile / methanol 40:60).
Yield: 4.32 kg (48% of theory.) Dissolved in 69.68 liters of acetonitrile / methanol 40:60 as a colorless fraction.
Enantiomeric purity:> 98.5% ee (HPLC, method D)
A sample is concentrated in vacuum to give: MS (EIPOS): m / z = 379 [M + H] +
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 1:05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H ), 5:37 (s, 1H), 6.60-6.84 (m, 2H), 7.14 (d, 1H), 7.28 (dd, 1H), 7:37 (d, 1H), 7:55 (s, 1H), 7.69 (s, 1H).
example 9
(4S) - 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (I)
Crystallization and Polymorphism setting
64.52 liters of a 6.2% solution of Example 8 in a mixture Acetonitiril / methanol 40:60 (equal 4.00 kg of compound 1) (1.2 .mu.m) via a filter cartridge and then concentrated at 250 mbar applicable so that the solution is still stirrable. It added 48 1 of ethanol denatured with toluene and distilled again at 250 mbar to stirrability from (Umdestillation on ethanol). They gave an additional 48 1 of ethanol denatured with toluene and then distilled at atmospheric pressure to a total volume of about 14 1 from (jacket temperature 98 ° C). The mixture was cooled via a ramp (4 hours) to 0 ° C, stirred for 2 hours at 0 ° C and filtered by the product from. It was washed twice with 4 1 of cold ethanol and then dried in vacuo at 50 ° C.
Yield: 3.64 kg (91% of theory.) Of a colorless, crystalline powder
Enantiomeric purity: "99% ee (HPLC method D); Retention times / RRT: (4S) - 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (1) ca. 11 min. RRT: 1.00; (4R) - 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (I) is about 9 min ,RRT: 0.82
Purity:> 99.8% (HPLC method B) RT: about 6.7 min.
Content: 99.9% (against an external standard)
specific rotation (chloroform, 589 nm, 19.7 ° C, c = 0.38600 g / 100 ml): - 148.8 °.
MS (EIPOS): m / z = 379 [M + H] +
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 1:05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H ), 5:37 (s, 1H), 6.60-6.84 (m, 2H), 7.14 (d, 1H), 7.28 (dd, 1H), 7:37 (d, 1H), 7:55 (s, 1H), 7.69 (s, 1H).
Melting point: 252 ° C (compound of formula (I) in crystalline form of modification I)
Physico-chemical characterization of compound of formula (I) in crystalline form of modification I
Compound of formula (I) melts in crystalline form of modification I at 252 ° C, ΔΗ = 95 -113 Jg 1 (heating rate 20 K min 1 , Figure 1).
A depression of the melting point was observed as a function of the heating rate.
The melting point decreases at a lower heating rate (eg 2 K min "1 ) because decomposition occurs. There were no other phase transitions. A mass loss of about 0.1% was observed up to a temperature of 175 ° C.


References
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel Herbst 2015 (German)
- Pitt, B; Anker, S. D.; Böhm, M; Gheorghiade, M; Køber, L; Krum, H; Maggioni, A. P.; Ponikowski, P; Voors, A. A.; Zannad, F; Nowack, C; Kim, S. Y.; Pieper, A; Kimmeskamp-Kirschbaum, N; Filippatos, G (2015). "Rationale and design of MinerAlocorticoid Receptor antagonist Tolerability Study-Heart Failure (ARTS-HF): A randomized study of finerenone vs. Eplerenone in patients who have worsening chronic heart failure with diabetes and/or chronic kidney disease". European Journal of Heart Failure 17 (2): 224–32.doi:10.1002/ejhf.218. PMID 25678098.
- Bakris, G. L.; Agarwal, R; Chan, J. C.; Cooper, M. E.; Gansevoort, R. T.; Haller, H; Remuzzi, G; Rossing, P; Schmieder, R. E.; Nowack, C; Kolkhof, P; Joseph, A; Pieper, A; Kimmeskamp-Kirschbaum, N; Ruilope, L. M.; Mineralocorticoid Receptor Antagonist Tolerability Study–Diabetic Nephropathy (ARTS-DN) Study Group (2015). "Effect of Finerenone on Albuminuria in Patients with Diabetic Nephropathy: A Randomized Clinical Trial". JAMA 314 (9): 884–94. doi:10.1001/jama.2015.10081. PMID 26325557.
 | |
| Systematic (IUPAC) name | |
|---|---|
| (4S)-4-(4-Cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1,6-naphthyridine-3-carboxamide | |
| Clinical data | |
| Legal status |
|
| Routes of administration | Oral |
| Identifiers | |
| CAS Number | 1050477-31-0 |
| ATC code | None |
| PubChem | CID 60150535 |
| ChemSpider | 28669387 |
| UNII | DE2O63YV8R |
| KEGG | D10633 |
| ChEMBL | CHEMBL2181927 |
| Synonyms | BAY 94-8862 |
| Chemical data | |
| Formula | C21H22N4O3 |
| Molar mass | 378.42 g/mol |
////Finerenone , BAYER, PHASE 3, BAY 94-8862
CCOC1=NC=C(C2=C1C(C(=C(N2)C)C(=O)N)C3=C(C=C(C=C3)C#N)OC)C
Urantide acts as selective and competitive urotensin-II (UT) receptor antagonist (pKB = 8.3), which blocks hU-II induced contractions in thoracic aorta ex vivo, and has no effect on noradrenaline or endothelin 1-induced contraction or on acetylcholine-induced relaxation. Urantide
ReplyDelete