
Trombopag series
1 Lusutrombopag
2 Avatrombopag
3 Selexipag
4 Totrombopag choline
5
6
7
1 LUSUTROMBOPAG
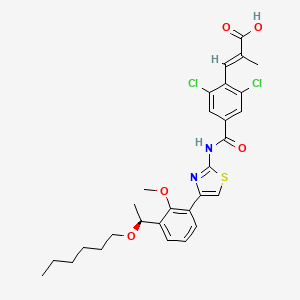
Lusutrombopag
(E)-3-[2,6-dichloro-4-[[4-[3-[(1S)-1-hexoxyethyl]-2-methoxyphenyl]-1,3-thiazol-2-yl]carbamoyl]phenyl]-2-methylprop-2-enoic acid
(S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid
(2E)-3-{2,6-Dichloro-4-[(4-{3-[(1S)-1-(hexyloxy)ethyl]-2-methoxyphenyl}-1,3-thiazol-2-yl)carbamoyl]phenyl}-2-methylacrylic acid
UNII 6LL5JFU42F, CAS 1110766-97-6,
D10476, MW591.546 , [US2010267783], MF C29H32Cl2N2O5S, S-888711
Shionogi & Co., Ltd., 塩野義製薬株式会社 INNOVATOROptically active compound (C-3B) Melting point: 142-145°C................EP2184279B1
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 - 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 - 3.37 (m, 2H), 1.69 (s, 3H), 1.18 - 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C).............EP2184279B1
Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C), NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 - 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 - 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs).........EP2184279B1
Thrombopoietin receptor agonist, Oral thrombopoietin (TPO) mimetic
- 24 Mar 2015 Shionogi plans a phase III trial in Thrombocytopenia (in patients with chronic liver disease) in USA (NCT02389621)
- 31 Dec 2014 Preregistration for Thrombocytopenia in Japan (PO)
- 08 Nov 2013 Phase II development is ongoing in the US and the Europe
In December 2014, an NDA was submitted in Japan. In May 2015, the drug was listed as being in phase III development for thrombocytopenia in the US and Europe.

The lusutrombopag, a low molecular-human thrombopoietin receptor agonist, its chemical formula, "(E) -3- [2,6-Dichloro-4- [4- [3 - [(S) -1-hexyloxyethyl] - 2-methoxyphenyl] -thiazol- 2-ylcarbamoyl] -phenyl] is a -2-methylacrylic acid ". lusutrombopag is represented by the following chemical structural formula.

Eltrombopag is represented by the following chemical structural formula.

Avatrombopag is represented by the following chemical structural formula.

Totrombopag choline is represented by the following chemical structural formula.


C 3B IS THE COMPD OF ROT (-) AND S, E FORM

Example 2
Synthesis
of
(R)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic
acid (C-3A) (not included in the present invention) and (S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic
acid (C-3B)
According to the same
method as in Example 1, an optically active compound (C-3A) and an
opticallly active compound (C-3B) were synthesized from
(RS)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic
acid (B-3) obtained in Reference Example 3.Optically active compound
(C-3A)Melting point: 139-141°C UNDESIRED
NMR
(DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8
Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 - 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5
Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 - 3.37 (m, 2H), 1.69
(s, 3H), 1.18 - 1.51 (m, 11H), 0.82 - 0.87 (m, 3H) Optical rotaion +4.5
degrees (DMSO, c = 1.001, 25°C)
Optically active compound (C-3B)Melting point: 142-145°C DESIRED
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 - 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 - 3.37 (m, 2H), 1.69 (s, 3H), 1.18 - 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)
Example 4: Synthesis of (C-3B)
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 - 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 - 3.37 (m, 2H), 1.69 (s, 3H), 1.18 - 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)
Example 4: Synthesis of (C-3B)

Using the same method as that of the first step of Example 3, the compound (17) was obtained from the compound (16) at a yield 77%.
- Optical rotation: -23.5 ± 0.6 degrees (CHCl3, c = 1.050, 21°C)
NMR (CDCl3) θ ppm: 1.49 (3H, d, J = 6.6 Hz), 2.33 (1H, brs), 3.88 (3H, s), 5.19 (1H, q, J = 6.4 Hz), 7.01 (1H, t, J = 7.9 Hz), 7.40 (1H, dd, J = 7.7 Hz, J = 1.1 Hz), 7.46 (1H, dd, J = 8.0 Hz, J = 1.4 Hz)
- Using the same method as that of the second step of Example 3, the compound (18) was obtained from the compound (17) at a yield of 96%.
Optical rotation: -29.8 ± 0.6 degrees (CHCl3, c = 1.055, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 - 1.4 (6H, m), 1.42 (3H, d, J = 6.5 Hz), 1.54 (2H, m), 3.29 (2H, m), 3.85 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 7.02 (1H, t, J = 7.9 Hz), 7.39 (1H, dd, J = 7.8 Hz, J = 1.7 Hz), 7.45 (1H, dd, J = 7.9 Hz, J = 1.7 Hz)
- Using the same method as that of the fourth step of Example 3, the compound (19) was obtained from the compound (18), subsequently according to the same method as that of the fourth step, the compound (20) was obtained.
- NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2-1.4 (6H, m), 1.45 (3H, d, J = 6.6 Hz), 1.55 (2H, m), 3.29 (2H, m), 3.78 (3H, s), 4.73 (2H, m), 4.80 (1H, q, J = 6.4 Hz), 7.24 (1H, t, J = 7.8Hz), 7.52 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.65 (1H, dd, J = 7.7 Hz, J = 1.8 Hz)
- Optical rotation: -4.2 ± 0.4 degrees (DMSO, c = 1.025, 21°C)
NMR (CDCl3) δ ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 - 1.3 (6H, m), 1.35 (3H, d, J = 6.5 Hz), 1.48 (2H, m), 3.25 (2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) - Fifth step: Synthesis of ethyl (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylate (21)
- Using the same method as that of the fifth step of Example 3, the compound (21) was obtained from the compound (20) at a yield of 94%.
Optical rotation: +4.7 ± 0.4 degrees (CHCl3, c = 1.07, 21°C)
NMR (CDCl3 ) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2 - 1.35 (6H, m), 1.38 (3H, t, J = 7.1
Hz), 1.44 (3H, d, J = 6.4 Hz), 1.57 (2H, m), 1.77 (3H, d, J = 1.4 Hz), 3.30 (2H, m), 3.59 (3H, s), 4.31 (2H, q, J = 7.1 Hz), 4.83 (1H, q, J = 6.4 Hz), 7.17 (1H, t, J = 7.7 Hz), 7.42 (1H, d, J = 1.7 Hz), 7.42 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.51 (1H, s), 7.67 (1H, dd, J = 7.6 Hz, J = 1.7 Hz), 7.89 (2H, s), 10.30 (1H, brs)
- Using the same method as that of the sixth step of Example 3, the compound (C-3B) was obtained from the compound (21) at a yield of 80%.Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 - 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 - 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) - Results of powder X-ray deffraction are shown in Fig. 5.
- Diffraction angle of main peak: 2θ = 17.8, 21.1, 22.5, 23.3, 24.1, and 24.4 degrees
WO2005014561/EP1655291A1
https://www.google.co.in/patents/EP1655291A1?cl=en
WO2014003155, claiming a composition comprising lusutrombopag, useful for treating thrombocytopenia.
https://www.google.co.in/patents/US20150148385?cl=en
.
WO 2015093586
Methods respectively for producing optically active compound having agonistic activity on thrombopoietin receptors and intermediate of said compound
(Step 1) Synthesis of compound (VII ') under a nitrogen atmosphere, it was dissolved compound 1 (2.00kg) in 1,2-dimethoxyethane (28.0kg). 25% LDA tetrahydrofuran - heptane - ethyl benzene solution (13.20kg) was added dropwise over 1 hour at -55 ℃, and stirred for 30 minutes. It was added dropwise over 40 minutes to 1,2-dimethoxyethane (3.0kg) solution of N- formyl morpholine (3.74kg) at -55 ℃, and stirred for 1 hour. 1,2-dimethoxyethane (3.0kg) solution of 2-phosphono-propanoic acid triethyl (3.74kg) was added dropwise over 45 minutes at 0 ℃, and stirred for 2 hours. 35% aqueous solution of sulfuric acid (15.8kg) was added dropwise over 40 minutes to the reaction solution. Water (16.0kg) was added and extracted. The resulting organic layer was washed with water (8.0kg), and the solvent was evaporated under reduced pressure. Acetonitrile (16.0kg) was added, and the mixture was stirred for 1 hour at 25 ℃, and the mixture was stirred and cooled to 0 ℃ 5 hours and 30 minutes. The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (16.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Over 1 hour and then cooled to 30 ℃, and the mixture was stirred for 45 minutes. Over 40 minutes and then cooled to 5 ℃, and the mixture was stirred for 3 hours.The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (13.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Furthermore, up to 30 ℃ over 1 hour and then cooled and stirred for 70 minutes. Over 30 minutes and then cooled to 5 ℃, and the mixture was stirred for 4 hours. I precipitated crystals were collected by filtration. Washed with 5 ℃ acetonitrile (3.2kg), and dried to give the compound (VII ') (1.63kg, 51.2% yield). NMR (CDCl 3 ) delta ppm: 8.07 (s, 2H), 7.47 (s, 1H), 4.32 (Q, 2H, J = 7.0 Hz), 1.79 (s, 3H), 1.38 (t, 3H, J = 7.0 Hz) Results of powder X-ray diffraction and I shown in Figure 1 and Table 3. [Table 3] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 8.1 ± 0.2 °, 16.3 ± 0.2 °, 19.2 ± 0.2 °, 20.0 ± 0. 2 °, the peak was observed at 24.8 ± 0.2 °, and 39.0 ± 0.2 ° degrees.
(Synthesis of Compound (XI '))
(Step 2) Synthesis of Compound 4 under a nitrogen atmosphere over Compound 3 (3.00kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (11.40kg) 1 hour at 25 ℃ in The dropped, and stirred for 2 hours. 1mol / L isopropylmagnesium chloride in tetrahydrofuran solution (0.56kg) was added at 25 ℃, and stirred for 2 hours. To the reaction mixture N- methoxymethyl -N- methylacetamide the (1.45kg) was added dropwise over at 25 ℃ 40 minutes, and stirred for 80 minutes. 7% hydrochloric acid (9.7kg) was added to the reaction mixture, and the mixture was extracted with toluene (11.0kg). The resulting organic layer twice with water (each 7.5kg) washed, the solvent was evaporated under reduced pressure to give Compound 4 (2.63kg). NMR (CDCl 3 ) delta ppm: 7.69 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.55 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.05 (t, 1H, J = 7.7 Hz), 3.88 (s, 3H), 2.64 (s, 3H) ppm:
(Step 3) Synthesis of Compound 5 Under a nitrogen atmosphere, chloro [(1S Compound 4 (2.63kg), 2S) -N- ( p- toluenesulfonyl) -1,2-diphenyl-ethane diamine] (p- cymene) ruthenium (II) (28.6g), it was added to tetrahydrofuran (1.3kg) and triethylamine (880.0g). Formic acid (570.0g) was added dropwise over 6 hours at 40 ℃, and stirred for 1 hour. In addition 3.5% hydrochloric acid (14.4kg) to the reaction mixture, and the mixture was extracted with toluene (13.0kg).The organic layer was washed with 3.5% hydrochloric acid (14.4kg) and water (7.5kg), the solvent was concentrated under reduced pressure to obtain a toluene solution of Compound 5 (4.44kg).
(Step 4) Synthesis of Compound 6 under a nitrogen atmosphere, it was a potassium hydroxide (6.03kg) was dissolved in water (6.0kg). To the solution, it added tetrabutylammonium bromide (182.0g) and toluene solution of Compound 5 (4.44kg). 1-bromo-hexane (2.79kg) was added dropwise over 1 hour at 60 ℃, and the mixture was stirred for 4 hours. And extracted by adding water (4.4kg) to the reaction solution. The resulting organic layer was filtered through powdered cellulose and extracted with toluene (3.0kg) and water (7.6kg) to the filtrate. The solvent it was evaporated under reduced pressure from the organic layer. Toluene operation of evaporated under reduced pressure and the solvent by the addition of a (7.8kg) was repeated five times to obtain a toluene solution of Compound 6 (10.0kg).
(Step 5) Synthesis of Compound 7 under a nitrogen atmosphere, magnesium powder (301.0g), in tetrahydrofuran (1.3kg), the compound in toluene (6.4kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (432.0g) 6 In addition of the toluene solution (0.50kg) at 30 ℃, and the mixture was stirred for 2 hours. Toluene solution of Compound 6 (9.50kg) was added dropwise over 3 hours at 50 ℃, and stirred for 2 hours. 1-bromo-hexane (746.0g) was added at 50 ℃, and the mixture was stirred for 1 hour. It was added dropwise over 1 hour at 5 ℃ toluene (5.3kg) solution of 2-chloro -N- methoxy -N- methyl-acetamide (1.78kg), and stirred for 1 hour. 3.7% hydrochloric acid (16.7kg) was added to the reaction mixture, and the mixture was extracted. The obtained organic layer was washed with water (15.0kg), and concentrated under reduced pressure to give a toluene solution of Compound 7 (8.25kg).
(Step 6) Synthesis of Compound (II ') under a nitrogen atmosphere, thiourea (1.03kg), in ethanol (1.2kg) and 65 ℃ toluene solution of compound 7 (8.25kg) in toluene (6.3kg) over 3 hours was added dropwise and stirred for 2 hours. The reaction solution was extracted by adding 0.7% hydrochloric acid (30.6kg), and washed twice with water (30.0kg). Ethanol in the organic layer (9.5kg), and extracted by addition of heptane (10.0kg) and 3.5% hydrochloric acid (5.9kg). The resulting aqueous layer with 4% hydrochloric acid (1.5kg) and ethanol (3.5kg) merged the aqueous layer was extracted from the organic layer, the ethanol was washed with heptane (10.0kg) (3.1kg) It was added. 8% aqueous sodium hydroxide (6.0kg) was added dropwise over at 5 ℃ 30 minutes, and stirred for 20 minutes. 8% aqueous sodium hydroxide (5.8kg) was added dropwise over a period at 5 ℃ 15 minutes.The precipitated crystals were collected by filtration, washed with 45% aqueous ethanol (10.9kg) and water (15.0kg) (crude crystals of Compound (II ')). The resulting crude crystals were dissolved in 50 ℃ in ethanol (8.1kg), over a period of 1 hour and then cooled to 10 ℃, and the mixture was stirred for 30 minutes. Water (10.0kg) over 2 hours was added dropwise and stirred for 30 minutes. The precipitated crystals were collected by filtration, washed with 50% aqueous ethanol (7.5kg) and water (10.0kg) (crystals of the compound after recrystallization from ethanol / water system (II ')). The resulting crystals were dissolved at 55 ℃ in toluene (1.6kg) and heptane (1.3kg), over 1 hour and cooled to 20 ℃, and stirred for 30 minutes. Heptane (6.3kg) over a period of 30 minutes was added dropwise and stirred for 15 minutes. The obtained crystals precipitated were collected by filtration, washed with a mixed solvent of toluene (0.3kg) and heptane (2.3kg), and dried to give compound (II ') (1.67kg, 44.5% yield) a (crystalline compound after recrystallization from toluene / heptane system (II ')).
NMR (CDCl 3 ) delta ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 - 1.3 (6H, M), 1.35 (3H, D, J = 6.5 Hz), 1.48 (2H, M), 3.25 ( 2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) it is shown in Figure 2 and Table 4 the results of powder X-ray diffraction. [Table 4] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 12.5 ± 0.2 °, 13.0 ± 0.2 °, 13.6 ± 0.2 °, 16.4 ± 0. 2 °, 23.0 ± 0.2 °, a peak was observed at 24.3 ± 0.2 ° degrees. Above, each of the compounds (II ') of the crude crystals, the ethanol / compound after recrystallization from water (II') crystals and toluene / heptane compound after recrystallization from (II ') crystallographic purity of the results of the , Fig. 3, I 4 and 5 as well as Table 5. [Table 5](HPLC was measured by the above method A.) As shown in the results of the above table, as compared to recrystallization from ethanol / water, recrystallized with toluene / heptane system, compounds having a high optical purity it is possible to manufacture a crystal of (II '). Next, the above-mentioned compound (II ') of the crude crystals, the ethanol / compound after recrystallization from water (II') crystals and toluene / heptane compound after recrystallization from (II ') results of crystals of HPLC of the respectively, Fig. 6, I 7 and 8 and Table 6. [Table 6] (units, .N.D shows the peak area of the (%). is, .HPLC to indicate not detected was measured by the above method B.) As shown in the results of Table, with ethanol / water system Compared to recrystallization, recrystallization from toluene / heptane system is found to be efficiently remove organic impurities A and organic impurities B.
(Step 7) Compound 'Synthesis of DMSO adduct of (VIII) Under a nitrogen atmosphere, the compound (II ') (1.50kg) and compound (VII') (1.43kg) in ethyl acetate (17.6kg) and triethylamine (1.09kg) were sequentially added, was dissolved.Diphenyl phosphorochloridate the (1.46kg) was added dropwise over 1 hour at 50 ℃, and the mixture was stirred for 3 hours. The reaction mixture was cooled to 25 ℃, after the addition of 2.6% hydrochloric acid (8.1kg), and extracted. The resulting organic layer to 6.3% aqueous solution of sodium hydroxide (3.2kg) and 14% aqueous sodium carbonate (5.2kg) was added and stirred for 20 minutes. Adjusted to pH7.5 with 8.3% hydrochloric acid and extracted. The organic layer it was washed with 4.8% sodium chloride aqueous solution (11.0kg). DMSO and (16.5kg) was added, and the mixture was concentrated under reduced pressure.DMSO and (5.8kg) was added, over a period at 40 ℃ 30 minutes was added dropwise water (0.9kg), and stirred for 1 hour. Over a period of 30 minutes, cooled to 25 ℃, and the mixture was stirred for 30 minutes. Over at 25 ℃ 30 minutes was added dropwise water (1.4kg), and the precipitated crystals were collected by filtration. After washing with 90% DMSO solution (10.0kg) and water (27.0kg), to obtain crystals of DMSO adduct and dried to Compound (VIII ') (2.98kg, 95.2% yield).
1H-NMR (CDCl 3 ) delta: 0.87 (t, J = 6.8 Hz, 3H), 1.20-1.34 (M, 6H), 1.37 (t, J = 7.1 Hz, 3H), 1.44 (D, J = 6.5 Hz , 3H), 1.52-1.59 (m, 2H), 1.77 (d, J = 1.3Hz, 3H), 2.62 (s, 6H), 3.28-3.34 (m, 2H), 3.59 (s, 3H), 4.31 ( q, J = 7.1Hz, 2H), 4.83 (q, J = 6.5Hz, 1H), 7.16 (t, J = 7.7Hz, 1H), 7.40-7.43 (m, 2H), 7.51 (s, 1H), 7.68 (dd, J = 7.7, 1.8Hz, 1H), 7.92 (d, J = 1.3Hz, 2H), 10.58 (s, 1H). The results of the powder X-ray diffraction and I are shown in Figure 9 and Table 7. [Table 7]
In the powder X-ray diffraction spectrum, diffraction angle (2θ): 5.2 ° ± 0.2 °, 7.0 ° ± 0.2 °, 8.7 ° ± 0.2 °, 10.5 ° ± 0.2 °, 12.3 ° ± 0.2 °, 14.0 ° ± 0.2 °, 15.8 ° ± 0.2 °, 19.3 ° ± 0.2 °, 22.5 ° peak was observed to ± 0.2 ° and 24.1 ° ± 0.2 °. TG / DTA analysis result it is shown in Figure 10. Then, each result of HPLC of concentrated dry solid and the above DMSO adduct crystals described in the following Reference Examples 1, 11 and 12, 13 and 14, and I are shown in Table 8. [Table 8] (unit, .HPLC showing peak areas of (%) was measured by the above methods C.) As shown in the results of the above Table, when compared with the extract, DMSO adduct of the compound (VIII ') The in the crystal, less residual organic impurities D, and it found to be about 56% removal.
(Step 8) under nitrogen atmosphere, DMSO adduct of the compound (VIII ') and (2.50kg) it was dissolved in ethanol (15.8kg). 24% sodium hydroxide aqueous solution (1.97kg) was added dropwise over a period at 45 ℃ 30 minutes to the solution and stirred for 3 hours. The reaction mixture was cooled to 25 ℃, water was added (20.0kg) and ethanol (7.8kg). 18% hydrochloric acid (2.61kg) was added dropwise over at 25 ℃ 30 minutes, followed by addition of seed crystals prepared according to the method described in Patent Document 23. After stirring for 3 hours and allowed to stand overnight. Thereafter, the precipitated crystals were collected by filtration, to give after washing with 50% aqueous ethanol solution (14.2kg), and dried to a compound (XI ') (1.99kg, 93.9% yield).
NMR (CDCl 3 ) delta ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 - 1.4 (6H, M), 1.48 (3H, D, J = 6.4 Hz), 1.52 - 1.64 (2H, M), 1.86 (3H, d, J = 1.4Hz), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) I is shown in Figure 15 the results of powder X-ray diffraction.
Patent Document 1: JP-A-10-72492 JP
Patent Document 2: WO 96/40750 pamphlet
Patent Document 3: JP-A-11-1477 JP
Patent Document 4: Japanese Unexamined Patent Publication No. 11-152276
Patent Document 5: International Publication No. 00/35446 pamphlet
Patent Document 6: JP-A-10-287634 JP
Patent Document 7: WO 01/07423 pamphlet
Patent Document 8: International Publication WO 01/53267 pamphlet
Patent Document 9: International Publication No. 02 / 059 099 pamphlet
Patent Document 10: International Publication No. 02/059100 pamphlet
Patent Document 11: International Publication No. 02/059100 pamphlet
Patent Document 12: International Publication No. 02/062775 pamphlet
Patent Document 13: International Publication No. 2003/062233 pamphlet
Patent Document 14: International Publication No. 2004/029049 pamphlet
Patent Document 15: International Publication No. 2005/007651 pamphlet
Patent Document 16: International Publication No. 2005/014561 pamphlet
Patent Document 17: JP 2005-47905 Japanese
patent Document 18: Japanese Patent Publication No. 2006-219480
Patent Document 19: Japanese Patent Publication No. 2006-219481
Patent Document 20: International Publication No. 2007/004038 pamphlet
Patent Document 21: International Publication No. 2007/036709 pamphlet
Patent Document 22: International Publication No. 2007/054783 pamphlet
Patent Document 23: International Publication No. 2009/017098 pamphlet
Non-Patent Document 1: Proceedings of the National Akademyi of Science of the United State of America (.... Proc Natl Acad Sci USA) 1992, Vol. 89, p 5640-5644.
Non-Patent Document 2: Journal of Organic (.. J. Org Chem) Chemistry 1984, Vol. 49, p 3856-3857.
Non-Patent Document 3: (.. J. Org Chem). Journal of Organic Chemistry, 1992, Vol. 57, p 6667-6669
Non-Patent Document 4:. Shinretto (Synlett) 2004 year Vol. 6, p 1092-1094
POSTER
| 101 | Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia Masami Takayama, Hajime Yamada, Hiroshi Takemoto, Takeshi Shiota, Yoshikazu Tanaka, Noriko Yamane, Kouji Takahashi, Naoki Oyabu, Kenji Kuwabara, Itsuki Oshima, Kenzo Koizumi, Hiroshi Yoshida, Ayumu Nogami, Tomomi Yamada, Yutaka Yoshida, Takami Murashi, Shinichiro Hara. |
| 101 - Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia Masami Takayama1, masami.takayama@shionogi.co.jp, Hajime Yamada3, Hiroshi Takemoto2, Takeshi Shiota2, Yoshikazu Tanaka2, Noriko Yamane2, Kouji Takahashi2, Naoki Oyabu3, Kenji Kuwabara3, Itsuki Oshima2, Kenzo Koizumi3, Hiroshi Yoshida3, Ayumu Nogami3, Tomomi Yamada3, Yutaka Yoshida3, Takami Murashi3, Shinichiro Hara2. (1) Department of Strategic Research Planning Offices, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan, (2) Department of Innovative Drug Discovery Research Laboratories, Shionogi & CO.,LTD, Toyonaka, Osaka 561-0825, Japan, (3) Department of Medicinal Research Laboratories, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan As a drug candidate of thrombocytopenia, Lusutrombopag (S-888711) is in Phase III clinical trial stage right now. It is been proven that Lusutrombopag (S-888711) is excellent property in safety and efficacy by clinical trials. In this meeting, we will present in detail about the history of drug discovery of Lusutrombopag.Because Lusutrombopag (S-888711) acts specifically to human TPO receptor, we prepared TPOR-Ki/Shi mice expressing a mouse-human chimeric TPOR for evaluating the efficacy. This TPOR-Ki/Shi mice worked very well as an evaluation model of drug efficacy, so we were able to select Lusutrombopag from many candidate compounds. In this meeting, we will present the results of the efficacy in TPOR-Ki/Shi mice of Lusutrombopag and the similar drug (Eltrombopag). Sunday, March 16, 2014 07:00 PM General Poster Session (07:00 PM - 10:00 PM) Location: Dallas Convention Center Room: Hall E Monday, March 17, 2014 08:00 PM Sci-Mix (08:00 PM - 10:00 PM) Location: Dallas Convention Center Room: Hall F |
2
Avatrombopag
Avatrombopag
AVATROMBOPAG; UNII-3H8GSZ4SQL; AKR-501; E5501; 570406-98-3; AS 1670542
| C29H34Cl2N6O3S2 | |
| Molecular Weight: | 649.65466 g/mol |
|---|
1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid,
1-(3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]pyridin-2-yl)piperidine-4-carboxylic acid,
1-[3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]-2-pyridyl]piperidine-4-carboxylic acid
4-Piperidinecarboxylic acid, 1-[3-chloro-5-[[[4-(4-chloro-2-thienyl)-5-(4-cyclohexyl-1- piperazinyl)-2-thiazolyl]amino]carbonyl]-2-pyridinyl]-
Phase III Clinical Trials
Drugs used in platelet disorders
Idiopathic thrombocytopenic purpura (ITP)
small-molecule thrombopoietin receptor (c-Mpl) agonist that stimulates platelet production
INNOVATOR: YAMANOUCHI PHARMACEUTICAL
DEVELOPER: Eisai

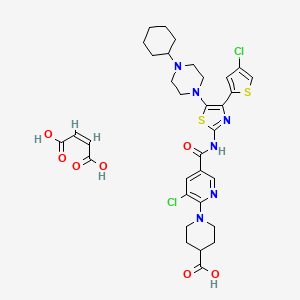
Avatrombopag maleate; UNII-GDW7M2P1IS; E5501 MALEATE; 677007-74-8; YM 477, AKR 501
| C33H38Cl2N6O7S2 | |
| Molecular Weight: | 765.72682 g/mol |
|---|
UNIIGDW7M2P1IS
(Z)-but-2-enedioic acid;1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acidINTRODUCTION

Avatrombopag is a thrombopoietin receptor (c-Mpl) agonist in phase III clinical evaluation at Eisai for the oral treatment of chronic immune thrombocytopenia (idiopathic thrombocytopenia purpura) and for the treatment of thrombocytopenia associated with liver diseases. Phase II studies are ongoing for the treatment of thrombocytopenia during antiviral therapy (inhibition and maintenance) with Interferon for hepatitis C.
The drug candidate may hold potential in treating thrombocytopenia of diverse etiologies, including idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia of myelodysplastic syndromes (MDS), in combination with or as a substitute for platelet transfusion.
AKR-501, a novel, small-molecule thrombopoietin mimetic being investigated for the treatment of thrombocytopenia. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture AKR-501. AKR-501 is an investigational thrombopoietin receptor agonist that, based on preclinical studies, increases platelet production by stimulating megakaryocytic proliferation and differentiation. Eisai is currently conducting Phase II clinical trials of AKR-501 in the United States as a potential treatment for idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia associated with liver diseases (TLD), and has confirmed proof of concept in the clinical studies for ITP. In addition, Eisai will explore the compound’s potential as a treatment for chemotherapy-induced thrombocytopenia (CIT).

E-5501 stimulates the production of thrombopoietin (TPO), a glycoprotein hormone that stimulates the production and differentiation of megakaryocytes, the bone marrow cells that fragment into large numbers of platelets. The drug candidate was originally developed at Yamanouchi, and development responsibilities were passed to AkaRx when it was formed in 2005 as a spin-off following the creation of Astellas Pharma subsequent to the merger of Yamanouchi Pharmaceutical and Fujisawa Healthcare.
In 2007, MGI Pharma was granted a license to E-5501 for the treatment of thrombocytopenia. Eisai eventually gained the rights to the product as results of its acquisition of MGI Pharma. In 2010, Eisai acquired AkaRx. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture E-5501. In 2011, orphan drug designation was assigned by the FDA for the treatment of idiopathic thrombocytopenic purpura.
E5501 (or AKR-501 or YM477) is a small molecule agonist c-Mpl, orally available. It is in clinical trials for the treatment of chronic idiopathic thrombocytopenic purpura (ITP). It acts as an agonist of the thrombopoietin receptor active orally, mimicking its biological effect. Thrombocytopenic purpura The is the idiopathic consequence of a low number of platelets (thrombocytopenia) of unknown cause. A very low platelets can even lead to purpura (bruises), or bleeding diathesis.
February 2012: A Phase III, multicenter, randomized, double-blind, controlled against placebo, parallel group, with an open-label extension phase to assess the efficacy and safety of combined oral E5501 to standard treatment for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia, is underway.
January 2010: Eisai Inc. announced its successful acquisition of the biopharmaceutical company, AkaRx Inc. Following this acquisition, AkaRx became a wholly owned subsidiary of Eisai Inc. Eisai now owns the worldwide exclusive rights to develop , marketing and manufacture AKR-501.
October 2009: Eisai Research Institute of Boston, Inc. (established in 1987) and Eisai Medical Research Inc. (established in 2002) were merged into Eisai Inc. 2005: AkaRx was founded as a spin-out of the merger of Yamanouchi Pharmaceutical Company Ltd. and Fujisawa Pharmaceutical Company Ltd. to form Astellas Pharma Inc. AKR-501 was discovered by Yamanouchi and was licensed to AkaRx as part of the foundation of the company in 2005.
In a Phase I trial in healthy volunteers, 10 mg of AKR-501 for 14 days, increased platelet count by 50%.AKR-501 was well tolerated in both studies, mono- and multi-dose. No adverse effects were reported, even at the highest doses.
……………………
Patent
WO 2004029049
Espacenet
Compound A is a compound of the present invention has the following chemical structure.

That is, compounds useful as a platelet 增多 agent according to the present invention A, as well as medicaments for the Compound A as an active ingredient, in particular increasing platelets agents and Z or thrombocytopenia treating agent.


………………
PATENT
WO 2003062233

……………………
JP 2014144916/WO 2013018362
https://www.google.co.in/patents/WO2013018362A1?cl=en
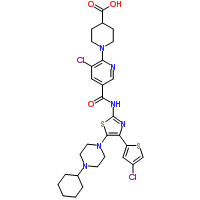
1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl}pyridin-2-yl)piperidine-4-carboxylic acid as expressed by the following chemical formula (hereinafter referred to as “Compound X”) and pharmaceutically acceptable salts are known to have excellent thrombocytosis effects (patent literature 1, patent literature 2).
[Formula 1]

Furthermore, patent literature 2 discloses a maleic acid salt of compound X that has endothermic peaks near 198 degree C and 271 degree C in thermo gravimetric analysis (hereinafter referred to as “maleic acid salt of compound X”). However, patent literature 2 neither discloses nor suggests that the maleic acid salt of compound X exhibits crystal polymorphism.
On the other hand, compounds exhibiting crystal polymorphism demonstrate entirely different effects regardless of being the same compound, because various physical properties including physicochemical properties differ depending on the crystalline form. In pharmaceutical products in particular, if compounds that have different functional effects are expected to have the same effect, a different functional effect than expected will occur, which is thought to induce unexpected circumstances, and therefore there is demand for supply of a drug substance with constant quality. Therefore, when a compound which has crystal polymorphism is used as a medicine, one type of crystal of that compound must always be constantly provided in order to ensure constant quality and constant effects that are required of the medicine.
Under the aforementioned conditions, from the perspective of supplying a drug substance for medicines, there is a need for compound X or crystals of pharmaceutically acceptable salts thereof, which can ensure constant quality and constant effects and which can be stably supplied in mass production such as industrial production or the like, as well as for establishment of a manufacturing method thereof.
International patent publication WO 03/062233 International patent publication WO 2004/029049
Preparation Example 1: Manufacture of Compound X Maleic Acid Salt B Type Crystal
310 mL of a 1 M aqueous solution of sodium hydroxide at room temperature was added to a mixture of 70.0 g of the ethyl ester of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid and 1.2 L of ethanol, the insoluble matter was filtered out, and then washed with 200 mL of ethanol. The reaction solution was stirred for 90 minutes at 60 degree C. After cooling to room temperature, 1.4 L of an aqueous solution containing 24.11 g of maleic acid was added to the solution obtained, and then the precipitate was collected by filtering.
The same operation was repeated and when combined with the previously obtained precipitate, 136.05 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid was obtained.
18.9 g of maleic acid and 2.1 L of 80% ethanol water were added to 88.90 g of the carboxylic acid obtained, and the solution was stirred for one hour at room temperature and for another hour at 100 degree C. After cooling to room temperature and further cooling with ice, the precipitated solid was filtered out to obtain 87.79 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
6.84 g of maleic acid was added to 231 g of the crude product containing the crude product obtained above and those manufactured in a similar manner, dissolved in 5.5 L of 80% ethanol water, and then the precipitated solid was collected by filtering to obtain 203 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 1: Manufacture of Compound X Maleic Acid Salt C Type Crystals (1)
1.52 L of ethanol, 0.38 L of water, and 15.7 g of maleic acid were added to 78.59 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, and heated while stirring. After cooling to room temperature and further cooling with ice, the precipitated solid was collected by filtering to obtain 71.60 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
296 mg of maleic acid was added to 10.0 g of the crude product obtained, dissolved in 60 mL of acetone, 60 mL of DMSO, and 30 mL of water, and then the precipitated solids were collected to obtain 8.41 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 2: Manufacture of Compound X Maleic Acid Salt C Type Crystals (2)
A mixture containing 80.1 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 580 mL of DMSO, 580 mL of acetone, 17.2 g of maleic acid, and 290 mL of water was stirred at 69 degree C. The insoluble matter was filtered out, washed with a mixture of 32 mL of DMSO, 32 mL of acetone, and 16 mL of water, and then the filtrate was cooled and the precipitate was collected by filtering. Washing was successively performed using 150 mL of water, 80 mL of acetone, 650 mL of water, and 80 mL of acetone, followed by drying, to obtain 70.66 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 3: Manufacture of Compound X Maleic Acid Salt C Type Crystals (3)
A mixture containing 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 100 L of DMSO, 100 L of acetone, 4.29 kg of maleic acid, and 50 L of water is stirred at 65 degree C, and then the insoluble matter is filtered out and washed with a mixture of 8 L of DMSO, 8 L of acetone, and 4 L of water, and then the filtrate is cooled, the precipitate is collected by filtering, successively washed using 40 L of acetone, 100 L of water, and 40 L of acetone, and then dried to obtain approximately 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
…………………………….
REFERENCES
Garabet, L.; Ghanima, W.; Lee, S.; Mowinckel, M.C.; Liebman, H.; Jonassen, C.M.; Bussel, J.; Sandset, P.M.
Thrombopoietin receptor agonists do no not cause coagulation activation: In patients with immune thrombocytopenia
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO311-MON
Terrault, N.; Hassanein, T.; Joshi, S.; Lake, J.R.; Sher, L.S.; Vargas, H.E.; McIntosh, J.W.; Tang, S.; Jenkins, T.
Once-daily oral avatrombopag (E5501) prior to elective surgical or diagnostic procedures in patients with chronic liver disease and thrombocytopenia: Results from a phase 2, randomized, double-blind, placebo-controlled study (study 202)
63rd Annu Meet Am Assoc Study Liver Dis (November 9-13, Boston) 2012, Abst
Thiophenyl Triazol-3-one Derivatives As Smooth Muscle relaxers: US6613786 (2003) Priority: US20010336865P, Nov. 2, 2001 (Bristol-Myers Squibb CO, US)
Preparation Of Avatrombopag: 2-Acylaminothiazole derivative or salt thereof: EP1466912 (2004) Priority: JP20020010413, 18 Jan. 2002 (Yamanouchi Pharma Co Ltd, Japan)
Synthesis And Use Of MSE Framework-Type Molecular Sieves: US2009318696 (2009) Priority: US20080214631 20 Jun. 2008 (Exxon Mobil, US).
5,6-Dichloro-Nicotinic Acid Production By Reacting 6-Hydroxy-Nicotinic Acid With Acid Chloride Reacting With Chlorine Products, Then With Acid Chloride And Hydrolysing Products: CH664754 (1988) Priority: CH19850002692, 25 Jun. 1985 (Lonza AG, Switzerland).
David J. Kuter, New Thrombopoietic Growth Factors, Lymphoma and Myeloma Clinical Journal Volume 9, Supplement 3, S347-S356
| WO2003062233A1 | 15 Jan 2003 | 31 Jul 2003 | Yamanouchi Pharma Co Ltd | 2-acylaminothiazole derivative or salt thereof |
| WO2004029049A1 | 29 Sep 2003 | 8 Apr 2004 | Yuuji Awamura | Novel salt of 2-acylaminothiazole derivative |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP2764866A1 | 4 Feb 2014 | 13 Aug 2014 | IP Gesellschaft für Management mbH | Inhibitors of nedd8-activating enzyme |
| Patent | Submitted | Granted |
|---|---|---|
| CANCER TREATMENT METHOD [US2011160130] | 2011-06-30 | |
| METHOD FOR STIMULATING PLATELET PRODUCTION [US2011166112] | 2011-07-07 | |
| COMPOSITIONS AND METHODS FOR INCREASING BLOOD PLATELET LEVELS IN HUMANS [US2011224226] | 2011-09-15 | |
| Method of treating viral diseases with combinations of TPO receptor agonist and anti-viral agents [US2012020923] | 2012-01-26 |
| Patent | Submitted | Granted |
|---|---|---|
| 2-Acylaminothiazole derivative or salt thereof [US7638536] | 2005-07-14 | 2009-12-29 |
| Compositions and methods for treating thrombocytopenia [US2007203153] | 2007-08-30 | |
| Novel Combinations [US2009304634] | 2009-12-10 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222329] | 2010-09-02 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222361] | 2010-09-02 | |
| Compositions and methods for increasing blood platelet levels in humans [US2008039475] | 2008-02-14 | |
| CANCER TREATMENT METHOD [US2009022814] | 2009-01-22 | |
| Compositions and methods for treating thrombocytopenia [US2010041668] | 2010-02-18 | |
| CANCER TREATMENT METHOD [US2010075928] | 2010-03-25 |
///////E 5501, AKR 501, Phase III, eisai, Avatrombopag, y 477, orphan drug, ym 477, AS 1670542, Yamanouchi Pharma Co Ltd, Japan
3
SELEXIPAG
FDA approves new orphan drug Uptravi (selexipag) to treat pulmonary arterial hypertension

12/22/2015
On December 21, the U.S. Food and Drug Administration approved
Uptravi (selexipag) tablets to treat adults with pulmonary arterial
hypertension (PAH), a chronic, progressive, and debilitating rare lung
disease that can lead to death or the need for transplantation.
December 22, 2015
On December 21, the U.S. Food and Drug Administration approved
Uptravi (selexipag) tablets to treat adults with pulmonary arterial
hypertension (PAH), a chronic, progressive, and debilitating rare lung
disease that can lead to death or the need for transplantation.
“Uptravi offers an additional treatment option for patients with pulmonary arterial hypertension,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “The FDA supports continued efforts to provide new treatment options for rare diseases.”
PAH is high blood pressure that occurs in the arteries that connect the heart to the lungs. It causes the right side of the heart to work harder than normal, which can lead to limitations on exercise ability and shortness of breath, among other more serious complications.
Uptravi belongs to a class of drugs called oral IP prostacyclin receptor agonists. The drug acts by relaxing muscles in the walls of blood vessels to dilate (open) blood vessels and decrease the elevated pressure in the vessels supplying blood to the lungs.
Uptravi’s safety and efficacy were established in a long-term clinical trial of 1,156 participants with PAH. Uptravi was shown to be effective in reducing hospitalization for PAH and reducing the risks of disease progression compared to placebo. Participants were exposed to Uptravi in this trial for a median duration of 1.4 years.
Common side effects observed in those treated with Uptravi in the trial include headache, diarrhea, jaw pain, nausea, muscle pain (myalgia), vomiting, pain in an extremity, and flushing.
Uptravi was granted orphan drug designation. Orphan drug designation provides incentives such as tax credits, user fee waivers, and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Uptravi is marketed by San Francisco-based Actelion Pharmaceuticals US, Inc.
“Uptravi offers an additional treatment option for patients with pulmonary arterial hypertension,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “The FDA supports continued efforts to provide new treatment options for rare diseases.”
PAH is high blood pressure that occurs in the arteries that connect the heart to the lungs. It causes the right side of the heart to work harder than normal, which can lead to limitations on exercise ability and shortness of breath, among other more serious complications.
Uptravi belongs to a class of drugs called oral IP prostacyclin receptor agonists. The drug acts by relaxing muscles in the walls of blood vessels to dilate (open) blood vessels and decrease the elevated pressure in the vessels supplying blood to the lungs.
Uptravi’s safety and efficacy were established in a long-term clinical trial of 1,156 participants with PAH. Uptravi was shown to be effective in reducing hospitalization for PAH and reducing the risks of disease progression compared to placebo. Participants were exposed to Uptravi in this trial for a median duration of 1.4 years.
Common side effects observed in those treated with Uptravi in the trial include headache, diarrhea, jaw pain, nausea, muscle pain (myalgia), vomiting, pain in an extremity, and flushing.
Uptravi was granted orphan drug designation. Orphan drug designation provides incentives such as tax credits, user fee waivers, and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Uptravi is marketed by San Francisco-based Actelion Pharmaceuticals US, Inc.
Selexipag, Uptravi
475086-01-2 CAS
(C26H32N4O4S, Mr = 496.6 g/mol)
A prostacyclin receptor (PGI2) agonist used to treat pulmonary arterial hypertension (PAH).
NIPPON SHINYAKU….INNOVATOR
Selexipag (brand name Uptravi) is a drug developed by Actelion for the treatment of pulmonary arterial hypertension (PAH). Selexipag and its active metabolite, ACT-333679 (MRE-269) (the free carboxylic acid), are agonists of the prostacyclin receptor, which leads to vasodilation in the pulmonary circulation.[1]
The US FDA granted it Orphan Drug status[2] (for PAH). It was approved by the U.S. FDA on 22 December 2015.[2]





PATENT
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer’s disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
PATENT
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a isthe drug
2a isthe drugN-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614

PATENT
WO 2011024874
Example 1 t- butylamine Form I crystal of the salt
Compound A (40 mg) with 0.5mL dimethoxyethane (hereinafter, referred to as. “DME”) was dissolved in, and t- butylamine (1.1 eq) were added, 25 1 ° C. at 8 it was stirred for hours. Thereafter, the reaction solution was added t- butyl methyl ether (1mL), at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, I-form crystals of t- butylamine salt ( 3 to afford 9.9mg). B Powder X-ray diffraction spectrum of type I crystal obtained t- butylamine salt using the apparatus shown in Figure 1.
Melting point: 152.5 ℃
elemental analysis (C 3 0 H 4 3 N 5 O 4 S + 0.0 3 H 2 as O)
calculated value (%) C: 6 3 .1 8 H: 7 . 6 1 N: 12 .2 8 measured value (%) C: 6 2. 8 5 H: 7 . 6 4 N: 12.52 1 H-NMR (DMSO-D 6 ): delta 8 .15 (s, 1H), 7 .55 – 7 . 8 0 (M, 2H), 7 .10- 7 . .45 (M, 10H), 4 7 . 0-4 8 5 (M, 1H), 3 . 6 6 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 3 (s, 3 H), 1.50-1. 7 5 (M, 4H), 1.2 3 (s, 9H), 1.22 (D, 6 H)
Example 2 I-form crystal of the potassium salt
Compound A tetrahydrofuran with (40mg) 12mL (hereinafter, referred to as. “THF”) was dissolved in, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added, 40 ℃ It was heated and stirred in for 15 minutes. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. After repeated two more times this step, at -20 ° C. 3 and held hours. The resulting precipitated crystals were collected by filtration under reduced pressure, and dried to obtain Form I crystal of the potassium salt. B Powder X-ray diffraction spectrum of type I crystal of the obtained potassium salt using the apparatus shown in Fig. 1 H-NMR (DMSO-D 6 ): delta 8 .14 (s, 1H), 7 .1 8 – 7 . 3 8 . (M, 10H), 4 7 . 2-4 8 4 (M, 1H) , 3 . 6 5 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 2 (s, 3 H), 1.55-1. 7 0 ( M, 4H), 1.2 3 (D, 6 H)
Example 3 II-form crystals of the potassium salt
Compound A with (40mg) was dissolved in THF and 12mL, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added and heated with stirring for 15 min at 40 ℃. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. This operation was repeated two more times, at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, after drying, 40 ℃, relative humidity 7 while 5% of thermo-hygrostat 7 left for days to give crystalline Form II of the potassium salt. B Powder X-ray diffraction spectrum of crystalline Form II of the resulting potassium salt using the apparatus Fig 3 is shown in.
Example 4 III type crystal of the potassium salt
Compound A , in addition to (100mg) acetonitrile (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (1.1 eq) was added and stirred for 200 minutes at 20 ℃. While stirring the mixture 7 after a heated stirring for 1 hour to 0 ° C., and then cooled to 10 ℃ over 10 hours. Further heated while the mixture 6 is heated to 0 ℃, t- butyl methyl ether (0. 3 after adding mL), cooled to 20 ℃ over 10 hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, III type crystal of the potassium salt ( 7 to afford 5mg). The powder X-ray diffraction spectrum of the type III crystal of the obtained potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, of about 7 endothermic peak was observed at around 4 ° C..
Elemental analysis (C 2 6 H 3 1 N 4 O 4 . SK + 0 7 8 H 2 as O)
calculated value (%) C: 5 6 .91 H: 5.9 8 N: 10.21
measured value (%) C: 5 6 . 6 1 H: 5.55 N:. 10 3 6
EXAMPLE 5 IV-type crystal of the potassium salt
Compound A , in addition to (50mg) and ethyl acetate (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (2.2 eq) was added and 2 at 20 ° C. 3 and stirred for hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried to obtain Form IV crystal of the potassium salt (41mg). The powder X-ray diffraction spectrum of crystalline Form IV of the resulting potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, an endothermic peak was observed at around approximately 91 ℃.

Selexipag (C26H32N4O4S, Mr = 496.6 g/mol) ist ein Diphenylpyrazin-Derivat. Es wird in der Leber zum aktiven Metaboliten ACT-333679 (MRE-269) biotransformiert. Selexipag unterscheidet sich strukturell von Prostazyklin und anderen Prostazylin-Rezeptor-Agonisten.

References
- 1 Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
- 2 New Drug Approved for Rare Lung Disorder. PPN. 23 Dec 2015 Has link to GRIPHON study results
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
| Patent | Submitted | Granted |
|---|---|---|
| Methods of identifying critically ill patients at increased risk of development of organ failure and compounds for the treatment hereof [US8877710] | 2009-12-30 | 2014-11-04 |
| Form-I crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide and method for producing the same [US8791122] | 2010-06-25 | 2014-07-29 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2015057325] | 2013-03-26 | 2015-02-26 |
| INHIBITION OF NEOVASCULARIZATION BY SIMULTANEOUS INHIBITION OF PROSTANOID IP AND EP4 RECEPTORS [US2014275200] | 2014-03-05 | 2014-09-18 |
| INHIBITION OF NEOVASCULARIZATION BY INHIBITION OF PROSTANOID IP RECEPTORS [US2014275238] | 2014-03-05 | 2014-09-18 |
| Fibrosis inhibitor [US8889693] | 2014-04-10 | 20 |
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound derivatives and medicines [US7205302] | 2004-05-27 | 2007-04-17 |
| METHODS OF IDENTIFYING CRITICALLY ILL PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ORGAN FAILURE AND COMPOUNDS FOR THE TREATMENT HEREOF [US2014322207] | 2014-07-11 | 2014-10-30 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2014329824] | 2014-07-18 | 2014-11-06 |
| Sustained Release Composition of Prostacyclin [US2014303245] | 2012-08-10 | 2014-10-09 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2013261177] | 2011-09-30 | 2013-10-03 |
| METHODS OF TREATMENT OF PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ISCHEMIC EVENTS AND COMPOUNDS HEREOF [US2013040898] | 2011-04-29 | 2013-02-14 |
| Substituted Diphenylpyrazine Derivatives [US2013005742] | 2010-08-06 | 2013-01-03 |
| USE OF FORM-I CRYSTAL OF 2–N-(METHYLSULFONYL)ACETAMIDE [US2014148469] | 2014-01-22 | 2014-05-29 |
| CRYSTALS OF 2- {4- [N- (5,6-DIPHENYLPYRAZIN-2-YL) -N-ISOPROPYLAMINO]BUTYLOXY}-N- (METHYLSULFONYL) ACETAMIDE [US2014155414] | 2014-01-22 | 2014-06-05 |
| PROSTACYCLIN AND ANALOGS THEREOF ADMINISTERED DURING SURGERY FOR PREVENTION AND TREATMENT OF CAPILLARY LEAKAGE [US2014044797] | 2012-03-30 | 2014-02-13 |
 |
|
| Names | |
|---|---|
| IUPAC name
2-{4-[(5,6-diphenylpyrazin-2-yl)(propan-2-yl)amino]butoxy}-N-(methanesulfonyl)acetamide
|
|
| Other names
ACT-293987, NS-304
|
|
| Identifiers | |
| 475086-01-2 |
|
| ChEMBL | ChEMBL238804 |
| ChemSpider | 8089417 |
| 7552 | |
| Jmol interactive 3D | Image |
| KEGG | D09994 |
| PubChem | 9913767 |
| UNII | P7T269PR6S |
| Properties | |
| C26H32N4O4S | |
| Molar mass | 496.6 g·mol−1 |
SEE........http://newdrugapprovals.org/2015/12/27/fda-approves-new-orphan-drug-uptravi-selexipag-to-treat-pulmonary-arterial-hypertension/
//////////
CC(C)N(CCCCOCC(=O)NS(=O)(=O)C)C1=CN=C(C(=N1)C2=CC=CC=C2)C3=CC=CC=C3
4
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He
was only in first standard in school when I was hit by a deadly one in a
million spine stroke called acute transverse mylitis, it made me 90%
paralysed and bound to a wheel chair, Now I keep him as my source of
inspiration and helping millions, thanks to millions of my readers who
keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने सेजिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
No comments:
Post a Comment