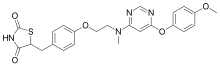
Enclomiphene citrate
NDA FILED Hypogonadism, Repros Therapeutics
An estrogen receptor (ER) antagonist potentially for treatment of hypogonadotropic hypogonadism.
ICI-46476; RMI-16289
CAS No.15690-57-0(free)
7599-79-3(Enclomiphene citrate)
| Molecular Weight | 598.08 |
| Formula | C26H28ClNO C6H8O7 C6H8O7 |
- Ethanamine, 2-[4-(2-chloro-1,2-diphenylethenyl)phenoxy]-N,N-diethyl-, (E)-, 2-hydroxy-1,2,3-propanetricarboxylate (1:1)
- Triethylamine, 2-[p-(2-chloro-1,2-diphenylvinyl)phenoxy]-, citrate (1:1), (E)-
- (E)-Clomiphene citrate
- Androxal
- Clomiphene B citrate
- Enclomid
- Enclomiphene citrate
- trans-Clomiphene citrate

Clomifene is a mixture of two geometric isomers, enclomifene (E-clomifene) and zuclomifene (Z-clomifene). These two isomers have been found to contribute to the mixed estrogenic and anti-estrogenic properties of clomifene.

Enclomifene
| 
Zuclomifene
|
PATENT
EXAMPLE 1
Preparation of trans-clomiphene citrate from
1- {4- [2-(Oiethylamino)ethoxy| phenylj-1 ,2-diphenylethanol
Dehydration
[0023] l-{4-[2-(Diethylamino)ethoxy]phenyl}-l,2-diphenylethanol (6) dissolved in ethanol containing an excess of hydrogen chloride was refluxed 3 hours at 50 °C. The solvent and excess hydrogen chloride were removed under vacuum and the residue was dissolved in dichloromethane. 2-{4-[(Z)-l,2-diphenylvinyl]phenoxy}-N,N- diethylethanaminium hydrogen chloride (7) was obtained.
Chlorination
The hydrochloride salt (7) solution obtained above was treated with 1.05 equivalents of N-chlorosuccinimide and stirred at room temperature for about 20 hours. Completion of the reaction was confirmed by HPLC. The hydrochloride salt was converted to the free base by addition of saturated aqueous bicarbonate solution. The mixture was stirred at room temperature for 30 minutes after which the phases were separated and the organic phase was evaporated in vacuo. 2-{4-[2-chloro-l,2- diphenylvinyl]phenoxy}-N,N-diethylethanamine (clomiphene -1.8:1 E:Z mixture) (8) was obtained.
Separation of clomiphene isomers
Clomiphene (8) obtained above is dissolved in methanol and racemic binaphthyl- phosphoric acid (BPA) is added under stirring. When the precipitate begins separating from the solution, stirring is stopped and the mixture is allowed to settle at room temperature for 2 hours. The precipitate is filtered, washed with methanol and ether and dried. Trans-clomiphene-BPA salt (3) is obtained.
The enclomiphene-BPA salt (3) obtained above is extracted with ethyl acetate and NH3 solution. To the organic solution washed with water and dried, citric acid dissolved in ethanol is added. The solution is allowed to settle for about one hour at room temperature; the precipitate is then filtered and dried under vacuum. The obtained precipitate, trans-clomiphene citrate (1) is dissolved in 2-butanone for storage.
EXAMPLE 2
Synthesis of Clomiphene Using a Single Solvent
Step 1 - Dehydration of l-i4-r2-(Diethylamino)ethoxy1phenyl|-l,2- diphenylefhanol to form 2-{4-[(Z)-l,2-diphenylvinyllphenoxy}-N,N-diethylethanaminium hydrogen sulfate (7) [0030] The synthesis route described in Example 1 utilized HC1 for the dehydration step and utilized ethanol at 50 °C as the solvent. Sulfuric acid was investigated as an alternative to HC1 for the dehydration step (as described in Example 1) in part due to the more favorable corrosion profile of sulfuric acid. Dichloromethane (methylene chloride) was investigated as an alternative solvent for the dehydration step as this would render removal of the ethanol solvent prior to the chlorination step unnecessary.
A 100 mL 3-neck round bottom flask, fitted with a temperature probe and a stir bar, was charged with l- {4-[2-(Diethylamino)ethoxy]phenyl}-l,2-diphenylethanol (6) (6.60 g, 16.9 mmol) and 66 mL (lxlO3 mmol) of methylene chloride to give a yellow solution which was cooled in an ice bath to 0 °C. Concentrated sulfuric acid (H2S04, 0.96 mL, 18.1 mmol) was added at a rate such that the internal temperature did not exceed 5 °C. Upon completion of the addition, the mixture was allowed to stir one hour at ambient temperature. Completion of the reaction was confirmed by high performance liquid chromatography (HPLC). The reaction resulted in 7.96 grams of 2- (4-[(Z)- 1 ,2- diphenylvinyl]phenoxy}-N,N-diethylethanaminium hydrogen sulfate (7), a yield of 100%. Thus, sulfuric acid was demonstrated to be a suitable acid for the dehydration step.
[0042] Using these HPLC conditions, starting material has a retention time of 3.30 min and product has a retention time of 4.05 min.
It was determined that removal of water produced by the dehydration reaction was important before performing the chlorination step. When ethanol is used as the solvent for this reaction, as in Example 1, the water is removed azeotropically upon removal of the ethanol. Several methods of drying the dichloromethane solution were attempted. Drying with MgS04 had a deleterious effect on the subsequent chlorination step, rendering the chlorination process very messy with a number of new impurities observed following HPLC analysis which were determined to be the corresponding chlorohydrins. On the other hand, a wash with brine was sufficient to remove enough water and had no deleterious effect on the chlorination step. Accordingly, the solution was stirred vigorously with brine (66 ml) for 30 minutes and then the phases were separated prior to chlorination step.
Step 2- Synthesis of 2-|4-r2-chloro-L2-diphenylvinyl1phenoxyl-N,N- diethylethanamine 8
The solution of 2-{4-[(Z)-l,2-diphenylvinyl]phenoxy}-N,N-diethylethanaminium hydrogen sulfate (7.94 grams) in methylene chloride obtained in step 1 is stirred at room temperature and treated with N-chlorosuccinimide (2.37 g, 17.7 mmol, 1.05 equivalents) in a single portion and left to stir at room temperature for 12 hours. The yellow solution became orange and then went back to yellow. After 12 hours, a sample was removed, concentrated and assayed by HPLC to confirm the extent of reaction. HPLC analysis revealed that the reaction had proceeded but not to completion. Accordingly, an additional 0.09 equivalents of N-chlorosuccinimide (203 mg, 1.52 mmol) was added and the solution stirred at room temperature for an additional 4 hours. The reaction was again assayed by HPLC which revealed that the reaction was near completion. Accordingly, an additional 0.09 equivalents of N-chlorosuccinimide (203 mg, 1.52 mmol) was added and the solution stirred for an additional 12 hours at room temperature. The reaction was again assayed by HPLC and an additional 0.058 equivalents of N-chlorosuccinimide (131 mg, 0.98 mmol) was added and the solution stirred for an additional 4 hours. HPLC indicated that the reaction was complete at that point. The reaction was carefully quenched by slow addition of 66 mL (600 mmol) of saturated aqueous sodium bicarbonate solution and the quenched mixture was stirred for 30 minutes at room temperature - the reaction mixture pH should be about 8-9 after addition of saturated aqueous sodium bicarbonate solution. The reaction yielded 6.86 grams of 2-{4-[2-chloro-l,2-diphenylvinyl]phenoxy}-N,N- diethylethanamine (8). The phases were separated and the organic phase was evaporated in vacuo. The resulting light brown oil was transferred to a tared amber bottle using a small volume of dichloromethane.
[0055] Using these HPLC conditions, the retention time of product is 15 minutes.
Chromatographic Separation of Clomiphene Isomers
Clomiphene (mixture of isomers) in free base form obtained by steps 1 and 2 is loaded onto a chromatographic column (e.g. batch high pressure chromatography or moving bed chromatography) using the same solvent as used in steps 1 and 2 (here DCM) in order to separate the cis- and trans-clomiphene isomers. Trans-clomiphene is preferably eluted using a solvent suitable for recrystallization.
PATENT
Indian (1978), IN 143841
PAPER
Separation of E- and Z-isomers of clomiphene citrate by high-performance liquid chromatography using methenamine as mobile phase modifier
Journal of Chromatography (1984), 298, (1), 172-4.
Journal of Chromatography (1984), 298, (1), 172-4.
PATENT
SYTHESIS

Patent
US2914562https://www.google.co.in/patents/US2914562
PATENT
US2914529
http://www.google.co.in/patents/US2914529
PAPER
J. Med. Chem.1967, 10, 84–86.
US2914562https://www.google.co.in/patents/US2914562
PATENT
US2914529
http://www.google.co.in/patents/US2914529
PAPER
J. Med. Chem.1967, 10, 84–86.

J. Org. Chem.1983, 48, 2782-2784.
PAPER
Chem Commun (London) 2015, 51(44): 9133
Chem. Commun., 2015, 51, 9133-9136
DOI: 10.1039/C5CC01968K
DOI: 10.1039/C5CC01968K
A transition-metal-free, ambient-pressure, and general methodology for carbonylative Suzuki coupling has been developed.

| CN103351304A * | Jul 1, 2013 | Oct 16, 2013 | 暨明医药科技(苏州)有限公司 | Synthesis method of clomiphene |
| US2914563 * | Aug 6, 1957 | Nov 24, 1959 | Wm S Merrell Co | Therapeutic composition |
| US3848030 * | Mar 10, 1972 | Nov 12, 1974 | Richardson Merrell Spa | Optical isomers of binaphthyl-phosphoric acids |
| US5681863 * | Dec 5, 1994 | Oct 28, 1997 | Merrell Pharmaceuticals Inc. | Non-metabolizable clomiphene analogs for treatment of tamoxifen-resistant tumors |
| Reference | ||
|---|---|---|
| 1 | * | RAO ET AL.: "Synthesis of carbon-14 labeled clomiphene.", JOUMAL OF LABELLED COMPOUNDS AND RADIOPHARMACEUTICALS, vol. 22, no. 3, 1985, pages 245 - 255, XP055180053, Retrieved from the Internet <URL:http://onlinelibrary. wiley .com/doi/10.1002/jlcr.2580220306/abstract> [retrieved on 20150504] |
//////////энкломифен, Enclomiphene citrate, إينكلوميفان , ICI-46476, RMI-16289, nda filed, Hypogonadism, Repros Therapeutics