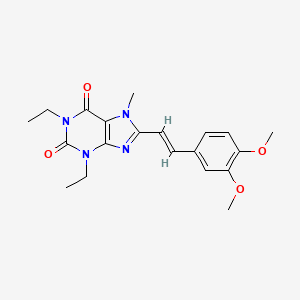
Istradefylline, KW-6002
(Nouriast®) Approved
A selective adenosine A2A receptor antagonist used to treat Parkinson's disease.
KW-6002
CAS No. 155270-99-8
Istradefylline; 155270-99-8; KW-6002; KW 6002; 8-[(E)-2-(3,4-Dimethoxyphenyl)ethenyl]-1,3-diethyl-7-methyl-purine-2,6 -dione; (E)-8-(3,4-Dimethoxystyryl)-1,3-diethyl-7-methyl-1H-purine-2,6(3H,7H)-dione;
| Molecular Formula: | C20H24N4O4 |
|---|---|
| Molecular Weight: | 384.42896 g/mol |
Istradefylline (KW-6002) is a selective antagonist at the A2A receptor. It has been found to be useful in the treatment of Parkinson's disease.[1] Istradefylline reduces dyskinesia resulting from long-term treatment with classical antiparkinson drugs such as levodopa. Istradefylline is an analog of caffeine.
Kyowa Hakko Kirin is developing istradefylline, a selective adenosine A2A receptor antagonist, for the once-daily oral treatment of Parkinson's disease (PD). Adenosine A2A receptors are considered to be present particularly in the basal ganglia of the brain; the degeneration or abnormality observed in PD is believed to occur in the basal ganglia, which is recognized to play a significant role in motor control.
Commercially available dopamine replacement therapies effectively treat the early motor symptoms of PD; however, these agents are associated with development of motor complications, limiting usefulness in late stages of the disease. Istradefylline is proposed to possess a clearly distinct action site from existing agents which act on dopamine metabolism or dopamine receptors. Kyowa Hakko Kirin has received approval for istradefylline in the adjunctive treatment of PD in Japan. A New Drug Application was filed in the USA, but the FDA issued a non-approvable letter in February 2008.
PATENT
US5484920A
PAPER

Scheme 1.

Synthesis
(E)-8-(3,4-Dimethoxystyryl)-1,3-diethyl-7-methyl-1H-purine-2,6(3H,7H)-dione (2)3
Synthesis of KW 6002 (2). Reagents and conditions: (i) acetic anhydride, 80 °C, 2 h, 83%; (ii) sodium nitrite, 50% acetic acid, 60 °C, 15 min, 86%; (iii) sodium dithionite, NH4OH solution (12.5% (w/v)), 60 °C, 30 min, 98%; (iv) SOCl2, toluene, 75 °C, 2 h, 97%; (v) pyridine, DCM, rt, 16 h, 66%; (vi) HMDS, cat. (NH4)2SO4, CH3CN, 160 °C, microwave, 5 h, 100% followed by (vii) MeI, K2CO3, DMF, rt, 2 h, 75%.
Synthesis
(E)-8-(3,4-Dimethoxystyryl)-1,3-diethyl-7-methyl-1H-purine-2,6(3H,7H)-dione (2)3
- J. Hockemeyer; J. C. Burbiel; C. E. Müller, J. Org. Chem. 2004, 69, 3308.
PATENT
Specific synthetic route is as follows:

the above reaction is a synthetic Parkinson's disease clinical drug KW-6002 against a yield of 83%.
Example 26 (a new synthetic method for anti-Parkinson's disease in clinical drug KW-6002):
In addition to use in place of 3,4-dimethoxy-styryl boronic acid (0.4mmol, i.e., in formula IV, R5 is 3,4_-dimethoxy-styryl) benzene boronic acid in Example 23 and 1,3 - two-ethyl-8-phenylthio-9-methyl-xanthine (0.4mmol, i.e., Formula I, R1 is methyl, R2 and R3 are ethyl, R4 is a phenyl group) in place of Example 23 in 1 , 3,9-trimethyl xanthine -8- phenylthio, the remaining steps in Example 23 to give a white solid, yield 83%, mp = 101~103 ° C I1H NMR (⑶CI3, 600MHz): δ 7.71 (d, J = 15.6Hz, 1H), 7.17 (dd, J = 8.2,1.9Hz, 1H), 7.07 (d, J = L 9Hz, 1H), 6
• 88 (d, J = 8.2Hz, 1H), 6.74 (d, J = 15.8Hz, 1H), 4.19 (q, J = 7Hz, 2H), 4.07 (q, J = 7Hz, 2H), 4.03 (s , 3H), 3.93 (s, 3H), 3.90 (s, 3H), 1.36 (t, J = 7Hz, 3H), 1.23 (t, J = 7Hz, 3H); 13C NMR (150MHz, CDCl3): 155.1, 150.8,150.4,150.2,149.2,148.2,138.2,128.6,121.2, 111.2,109.5,109.3,108.0,56.0,55.9,38.4,36.3,31.5,13.4,13.4; HRMS: calcd for C20H25N4O4 (M + H) +385.187
6, Found385.1879. It indicates that the white solid was 8- (3,4-dimethoxy-styryl) structural formula shown KW-6002 (E) -1,3_ diethyl-7-methylxanthine.

In contrast, KW-6002 is a new drug to treat Parkinson's disease developed by Kyowa Hakko in Japan, Japan and the United States is currently the second phase of clinical trials. Literature (. J.Hockemeyer, JCBurbiel andC.E.Muller, J.0rg.Chem, 2004,69,3308) through the following synthetic route:

The synthetic route requires five steps, with a total yield of 33%, and there is the use of environmentally unfriendly halogenated solvent methylene chloride, the reaction requires high pressure high temperature (170~180 ° C) and other shortcomings. By comparison, the present invention starting from 8- phenylthio xanthine coupling reaction catalyzed by palladium simple, a yield of 83% was synthesized KW6002, it is currently the most efficient synthesis route KW-6002's. In particular, the multi-step synthesis route to avoid the complex operation of the reactor, but under relatively mild conditions (60 ° C) conduct, simple operation, suitable for scale synthesis.
PATENT
itraconazole theophylline (Istradefylline, KW6002), the chemical name 8 - [(E) -2- (3, 4- dimethoxyphenyl) ethenyl] -1,3-diethyl -7 - methyl-purine-2,6-dione, CAS number: 155270-99-8, structural formula shown below.

itraconazole Theophylline is a selective adenosine A2a receptor antagonist, by changing the activity of neurons in Parkinson's disease patients to improve motor function, for the treatment of Parkinson's disease and Parkinson's disease improve early dyskinesia.
The invention and JPH0940652A European Patent 0,590,919 discloses a method for preparing itraconazole and theophylline. WO 2004/099207 published good solubility stability of a particle size of less than 50 micrometers 8 - [(E) -2- (3, 4- dimethoxyphenyl) ethenyl] -1,3- diethyl-7-methyl-purine-2,6-dione crystallites.
Example 1 Preparation of theophylline itraconazole Example

ships equipped with a mechanical stirrer, a thermometer, a 2L 4-neck flask was added 30g8 - [(E) -2- (3, 4- dimethoxyphenyl) ethenyl] -1,3-diethyl- -7- hydrogen - purine-2,6-dione (Intermediate A), 400mL N, N- dimethylformamide and 15g of potassium carbonate, and 25g of methyl iodide and heated to 80 ° C after the reaction was stirred 8h, added 200mL water, cooled to room temperature, and stirring was continued crystallization 2h. The resulting suspension was suction filtered, washed with water after the cake was 800mL sash, 50 ° C under blast drying 24h, 32g give a pale yellow solid, for each polymorph of itraconazole theophylline preparation example the following examples.
References
- Peter A. LeWitt, MD, M. Guttman, James W. Tetrud, MD, Paul J. Tuite, MD, Akihisa Mori, PhD, Philip Chaikin, PharmD, MD, Neil M. Sussman, MD (2008). "Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces off time in Parkinson's disease: A double-blind, randomized, multicenter clinical trial (6002-US-005)". Annals of Neurology 63 (3): 295–302. doi:10.1002/ana.21315. PMID 18306243.
1. EP0590919A1.
2. US5484920A.
3. US5543415A.
4. J. Org. Chem. 2004, 69, 3308-3318.
5. Bioorg. Med. Chem. Lett. 1997, 7, 2349-2352.
6. Bioorgan. Med. Chem. 2003, 11, 1299-1310.
7. Bioorg. Med. Chem. Lett. 2013, 23, 3427-3433.
8. Chinese Journal of Pharmaceuticals 2010, 41, 241-243.
9. JP0940652A.
10. Org. Biomo. Chem. 2010, 8, 4155-4157.
2. US5484920A.
3. US5543415A.
4. J. Org. Chem. 2004, 69, 3308-3318.
5. Bioorg. Med. Chem. Lett. 1997, 7, 2349-2352.
6. Bioorgan. Med. Chem. 2003, 11, 1299-1310.
7. Bioorg. Med. Chem. Lett. 2013, 23, 3427-3433.
8. Chinese Journal of Pharmaceuticals 2010, 41, 241-243.
9. JP0940652A.
10. Org. Biomo. Chem. 2010, 8, 4155-4157.
1. Chem. Commun. 2012, 48, 2864-2866.
2. CN103254194A.
2. CN103254194A.
| CN104744464A * | Nov 15, 2013 | Jul 1, 2015 | 南京华威医药科技开发有限公司 | Istradefylline crystal forms |
Istradefylline Systematic (IUPAC) name 8-[(E)-2-(3,4-dimethoxyphenyl)vinyl]-1,3-diethyl-7-methyl-3,7-dihydro-1H-purine-2,6-dione Identifiers CAS Number 155270-99-8 
ATC code none PubChem CID 5311037 IUPHAR/BPS 5608 ChemSpider 4470574 UNII 2GZ0LIK7T4 KEGG D04641 ChEMBL CHEMBL431770 Chemical data Formula C20H24N4O4 Molar mass 384.429 g/mol

//////Istradefylline, KW-6002, Nouriast®, Approved, A selective adenosine A2A receptor antagonist, Parkinson's disease,
O=C2N(c1nc(n(c1C(=O)N2CC)C)\C=C\c3ccc(OC)c(OC)c3)CC