PAROXETINE

- The reaction of 4- (4-fluorophenyl) -1-methyl-1,2,3,6-tetrahydropyridine (I) with formaldehyde and H2SO4 gives racemic 4- (4-fluorophenyl) -3- (hydroxymethyl) -1-methyl-1,2,3,6-tetrahydropyridine (II), which is submitted to optical resolution with (-) - dibenzoyltartaric acid, yielding the 3 (S) -enantiomer (III) The reduction of (III). with H2 over Pd / C in ethanol affords trans- (3S, 4R) -4- (4-fluorophenyl) -3- (hydroxymethyl) -1-methylpiperidine (IV), which is treated with SOCl2 to provide the corresponding chloromethyl derivative ( V). The condensation of (V) with 1,3-benzodioxol-5-ol (VI) by means of NaOMe in methanol furnishes trans- (3S, 4R) -3- (1,3-benzodioxol-5-yloxymethyl) -4- (4-fluorophenyl) -1-methylpiperidine (VII), Which is Condensed with phenyl chloroformate (VIII) in DICHLOROMETHANE to afford the phenyl carbamate (IX). Finally, the phenoxycarbonyl group of (IX) is removed by treatment with KOH in refluxing methylcellosolve or in refluxing toluene. Alternatively, the reaction of methylpiperidine (VII) with ethyl chloroformate (X) in toluene gives the ethyl carbamate (XI), which is finally treated with KOH in refluxing ethanol / water in order to eliminate its Ethoxycarbonyl group. Alternatively, the Reaction of methylpiperidine (VII) with Vinyl chloroformate (XII) in DICHLOROMETHANE Gives the Vinyl carbamate (XIII), Which is finally Treated with Dry HCl Gas in refluxing DICHLOROMETHANE / methanol in Order to Eliminate ITS vinyloxycarbonyl group .
Process development and optimisation studies for the manufacturing process of N-methyl paroxetine was done by applying Quality by Design (QbD) principles. A normal operating range was defined for each process parameter by technique of design of experiments and knowledge of process chemistry to ensure consistent high quality of advanced intermediate and control of impurities. Initial challenges faced during early process development, were overcome by novel approach using polar aprotic solvents like sulpholane. Finally DMSO was used to improve overall impurity profile significantly.We have demonstrated a “ right first time” approach to avoid failures and bitter surprises during scale up in manufacturing plant.
INTRODUCTION
Process development and scale- up of commercially important active pharmaceutical ingredients( APIs) is an interesting and challenging task to an organic chemist in the pharmaceutical field. However, due to ever rising cost competition and requirements by formulation colleagues in stipulated time frame, it becomes an ever increasing daunting task. If process development is done in certain logical steps and in a systematic manner, it could be a satisfying experience with respect to timely delivery of APIs and launching of commercially viable new generic molecules. Process development by the traditional “one change at a time” paradigm leads to a large number of experiments thus consuming resources of an organisation and time to market is delayed considerably. To reduce the number of experiments required for effective process development “design of experiments” strategies and software programs like Stavex®, Dynochem®, Modde®( from Umetrics, Sweden) [1-7] are being used by process development chemists worldwide. Access to these softwares, training and implementation are still a distant dream for organic/ process development chemist working in small to medium-sized Indian pharmaceutical organisation. Hence systematic and logical thinking towards designing a manufacturing process is very much an essential factor. Imagination of step-by-step unit operations during large scale manufacturing and “what if certain thing goes wrong?” kind of thinking would give positive inputs while designing a robust process. To answer such obvious questions, leads to negative experiments and data generation to define the “normal operating range” of the process and also to understand the “ edge of potential failure” concisely.
In this paper, we would like to elaborate more on simple strategies during API process development [8] and how process chemist can benefit from such simple experiments, in a minimum number of laboratory experiments, thus saving on resources and environmental aspects as well. Each process parameter can be challenged to “lower than” and “higher than” the limit of a fixed parameter and experiments can be envisaged accordingly. Analytical data generated by such experimentation throws more light on the problem and an in-depth knowledge of the process could be sought before actually going for scale-up in the manufacturing facility. Generation and interpretation of analytical data, in-process control checks and intermediate analysis reports gives more assurance towards achieving successful scale-up campaigns, yields and quality of APIs.
Initial challenges like glassy nature of mesylate intermediate, which could not be removed comfortably from reaction flask. It was triturated with cyclohexane to obtain material in the form of powder. Later on this isolation was removed and mesylate intermediate was taken “in situ” and condensed with sesamol. In condensation step, reaction rate was significantly improved to 2-3 hours by novel approach of using polar aprotic solvent like sulpholane. Impurity carry over from N-methyl Paroxetine intermediate was observed and hence finally DMSO solvent was used in condensation step to improve overall impurity profile significantly to achieve quality as per ICH guidelines.
MATERIALS AND METHODS
Process Development Strategies
Among the various strategies being developed by process chemists, we would like to summarize a few of them which are commonly adopted worldwide. In a chemical reaction there are starting materials, reagents, catalysts and solvents, which are necessary raw materials required to produce an API. To monitor the progress of reaction for its completion, various sophisticated modern analytical tools like Thin layer chromatography(TLC), High pressure liquid chromatography (HPLC), Gas chromatography (GC), Proton nuclear magnetic resonance (1HNMR) are used. To isolate the intermediate/product various purification techniques like solvent extraction, precipitation by addition of solvent in which material has less solubility also called as an antisolvent, recrystallisation in single or mixed solvents, ion exchange resins, pH adjustments and filtration etc. are employed routinely. Purification by column chromatography is also an important technique for achieving desired purity of final API as per regulatory guidelines. For removal of solvent from wet material, techniques like drying under vaccum in vacuum tray dryers, in rotary cone dryers, in fluidised bed dryers etc. are employed. Finally in order to achieve the desired particle size equipments like multi-mills, ball mills, pin mills, microniser/air jet mills, spray dryers, lyophilisers are used. Hence proper equipment selection for the desired unit operations also constitutes the robustness of a manufacturing process. Identification of critical process parameters with respect to quality and safety is also done during designing the process.
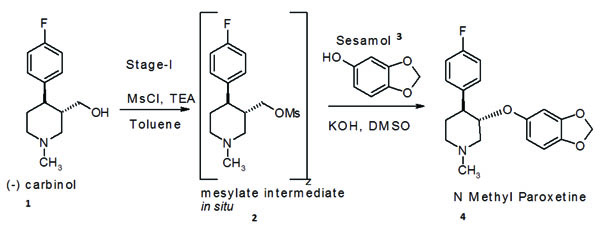
Paroxetine hydrochloride,trans(−)-3-(1,3-benzodioxol-5-yloxy)methyl-4-(4-fluorophenyl) piperidine, is an antidepressant [9] developed by SmithKline Beecham and also used in treatment of obsessive compulsive disorder under brand name Paxil®. We will take as an example the development of a process for an advanced (N-1) stage intermediate required for manufacture of paroxetine hydrochloride hemihydrate. N-methyl paroxetine is synthesised by condensation of chirally pure (-)(3S,4R)-4-(4-fluorophenyl)-1-methylpiperidin-3-ylmethanol i.e.(-) carbinol(1) with 3,4 dihydroxymethylene phenol i.e. sesamol (3)in a polar organic solvent using an inorganic base Scheme (1).
Scheme. (1)
Scheme. (1)
Brief Process Development History
(-) carbinol (1) was first converted into an in situ generated “mesylate intermediate (2)” using triethylamine (TEA) as a HCl scavenger and toluene as solvent. This was then condensed with sesame in the next step of to obtain N-methyl paroxetine, using the novel solvent sulpholane and polar solvents like DMSO, N-methyl pyrrolidone, dimethyl acetamide, 2-ethoxy ethanol or methanol. N-methyl paroxetine obtained by this method was far more superior in quality than obtained by using prior art methods [10-27] and the overall yield was in the range of 80% to 85% with respect to the starting (-) carbinol.
It was also observed that the mesylation reaction using triethylamine (TEA) as base was exothermic ( ΔHR = 1117KJ/Kg, ΔT adiabatic = 67°C) in nature. Hence to avoid run away situations in large scale manufacture, it was necessary to carry out this reaction at low temperature and the exotherm was controlled by slow addition of the methanesulphonyl chloride reagent. This can be better controlled by diluting methanesulphonyl chloride in toluene solvent.
For the convenience of experiments, it was necessary to isolate the mesylate intermediate as a solid powder. Hence the thick oily mesylate intermediate was triturated, with cyclohexane and the solid powder obtained was filtered on a Buchner funnel and could be dried easily under vacuum. HPLC analysis indicated a purity exceeding 95% and the material was not hygroscopic and it was stable at room temperature storage conditions for prolonged time. In general, mesylate intermediates are unstable and not isolated for this reason, but in this case, we would like to emphasize that the mesylate intermediate was quite stable at room temperature and could be isolated as a solid powder by trituration in cyclohexane as mentioned above. It was also possible to analyse the material without any difficulty by HPLC using aqueous buffer solutions.
The condensation reaction with sesamol using sulpholane solvent generated unknown impurities in the N-methyl paroxetine intermediate [28]. They were converted (carry over) to unknown impurities in the final Paroxetine API. Therefore impurities must be removed at the N-methyl paroxetine stage to avoid purification of final API. When purified N-methyl paroxetine was converted to the final API, it complied with USP specifications and criteria [29]. However, the cost of manufacturing increased by almost 20-30%, thus the process was not commercially viable. Hence to avoid purification of N-methyl paroxetine, DMSO was selected as a solvent for reaction and process parameters were optimised accordingly. Analytical data were generated for each parameter change in the process and compiled to better understand the manufacturing controls during the execution of batches.
Process flow chart: In order to understand the process flow, the following process flow diagram was drawn depicting the major unit operations and their sequence as per the design of the process. Fig. (1). Various experiments were designed to study the negative impact on quality of the intermediate and yield of the process. After having set down the process for the “mesylate intermediate”, it was taken “in situ” and the subsequent condensation step with sesamol was further optimised.
Fig. (1) Process flow diagram.

RESULTS AND DISCUSSION
Choice of reagent: It is necessary to convert a primary hydroxy group into a good leaving group in order to condense the (-) carbinol moiety with sesamol as per SN2 reaction mechanism. This can be achieved by employing either p-toluene sulphonyl chloride or methane sulphonyl chloride. From the perspective of atom economy, methane sulphonyl chloride was selected, which is also commercially cheaper reagent and readily available.
Reagent stoichiometry: Ideally, 1 mole equivalent of methanesulphonyl chloride MsCl converts (-) carbinol into mesylate, but practically a larger amount of reagent is required due to trace levels of moisture in the raw materials and solvents, which hydrolyses methanesulphonyl chloride to methane sulphonic acid, thus impeding the desired reaction from going to completion. Hence an additional quantity of reagent (approximately 5mole % to 10 mole%) is required for completion of reaction and conversion into mesylate intermediate. The reaction progress was conveniently monitored by TLC. Initial experiments were carried out starting with 1.1-1.2 mole equivalents of Methane sulphonyl chloride. It was observed that reaction was not complete with 1.1 -1.2 moles equivalent and starting material (-) carbinol remained unreacted in the range of 1% to 6% . Table 1.
Table 1: Deciding Reagent Stoichiometry
| Expt. No. | Solvent Volumes | Process Set Point | MsCl Moles eq | HPLC Results | Comments |
|---|---|---|---|---|---|
| Expt No. 1 | 10 volume | 1.25 - 1.35 mole equivalents of MsCl. | 1.1 | 98.09% Mesylate, 1.23 % carbinol | Yield: 97.9% |
| Expt No. 2 | 10 volume | 2.0 | 98 A2% Mesylate, 0.15 % carbinol | Yield: 57.7% | |
| Expt No. 3 | 10 volume | 1.0 | 91.97% Mesylate, 5.78 % carbinol | Yield: 94.81% | |
| Expt No. 4 | 10 volume | 1.5 | 98.96% Mesylate, 0.7% carbinol | Yield: 90.37% |
During the optimization of the process, reactions were monitored by HPLC for more accuracy of the results. It was observed that in order to convert (-) carbinol to mesylate, 1.25-1.35 mole equivalents of methane sulphonyl chloride reagent are required. Since the reaction is exothermic, the reagent was diluted with 2 volumes of toluene with respect to the input of (-) carbinol and added slowly into the reaction mixture to maintain a reaction temperature below 20°C. Higher mole equivalents of MsCl are not advisable as it affects the yield of the reaction.
Choice of Solvent [31] and Volumes
Initial trials were done using dichloromethane and toluene solvent for the mesylation reaction. International conference on harmonisation (ICH) guidelines[30] do not recommend chlorinated solvents like dichloromethane due to their toxic potential and adverse side effects on occupational health of shop floor personnel and patients. Recovery of chlorinated solvents is a tedious task and huge losses are incurred during recovery of solvents. Therefore use of chlorinated solvents becomes a costly affair. Toluene, on other hand, is cheaper and upto 90% of this solvent can be recovered and reused. Hence toluene solvent was selected.
Solvent volume ratio: The solvent-substrate volume ratio was optimized to 10 volumes with respect to starting material. There is a formation of TEA.HCI salt due to HCI scavenging by TEA. This results in the reaction mass becoming a thick paste and so as to ensure homogeneity of the reaction mass, 10 volumes of toluene are necessary. If smaller volumes are used i.e. 5 volumes of toluene, the quality of the mesylate intermediate is compromised. On the other hand, if larger volumes are used i.e. 15 volumes the quality of mesylate is not affected.
Temperature of reaction: As noted previously, the temperature of the reaction mixture increases due to the exothermic nature of the reaction and therefore must be controlled by slow addition of methane sulphonyl chloride. This protocol is a critical step from the safety point of view. Addition of methane sulphonyl chloride also can be controlled by diluting the reagent with toluene solvent. This also gives a better control on the temperature rise and hence the exothermicity of the reaction. The temperature can be controlled up-to a maximum of 20°C without affecting the quality of the mesylate intermediate.
Triethyl amine stoichiometry: Prio art patents and references describe the use of pyridine as an HCl scavenger. Use of pyridine is banned by environmental protection agencies [31] because of its inherent toxicity [31], adverse side effects due to exposure and environmental hazard. Hence the safer and cheaper triethylamine was chosen as an alternative for the intended application.Ideally 1 mole equivalent of triethylamine should convert (-) carbinol into mesylate, but practically 2.2 to 2.35 molar equivalents of reagent is required due to hydrolysis of methanesulphonyl chloride to methane sulphonic acid, as described in the reagent stoichiometry section. Initial experiments were carried out starting with 2.0-3.0 mole equivalents of triethylamine. It was observed that more impurities were formed when lesser i.e.1.5 moles equivalents of triethylamine were used. Higher equivalents of TEA did not affect the quality of mesylate intermediate. Hence 2.2 -2.35 mole equivalents of TEA were selected Table 2.
Table 2: Deciding TEA Stoichiometry
| Expt No. | Moles of MsCl | Process Set Point | TEA Moles eq | HPLC Results | Comments |
|---|---|---|---|---|---|
| Expt No. 5 | 1.3 | 2.2 to 2.35 mole equivalents of TEA. | 1.5 | 93.89% Mesylate, 5.81 % UK Yield: 87.40% | TEA qty should not be less than optimised qty. |
| Expt No. 6 | 1.3 | 3.0 | 99.18% Mesylate, 0.81 % carbinol Yield: 93.33% | No adverse effect on quality of mesylate when excess of TEA was used | |
| Expt No. 7 | 1.3 | 2.1 | 94.66% Mesylate, 4.85 % UK | Yield: 93.33% | |
| Expt No. 8 | 1.3 | 2.3 | 99.22% Mesylate, 0.77% carbinol | Yield: 90.37% |
In situ intermediate: After having set the optimum parameters for “ mesylate intermediate” i.e. 1.25-1.35 mole equivalents of methanesulphonyl chloride, 2.2 to 2.35 mole equivalents of Triethylamine, 10 volumes of toluene solvent and 0°C to 10°C reaction temperature, we decided not to isolate “ mesylate intermediate” but it was kept “in situ” and further optimisation experiments for the reaction conditions/parameters for the subsequent condensation reaction with sesamol to obtain N-methyl paroxetine( NMP) were carried out.
Time Required for Completion of Reaction:
Mesylation reactions are very fast on small scale [32-38]. However, when batches were scaled up to100-200 g scale and monitored by HPLC for completion time, it was observed that after addition of methane sulphonyl chloride reagent, 2-3 hours time was necessary for complete conversion to the mesylate intermediate.
Reaction monitoring HPLC data showed that (-) carbinol remained unreacted even after stirring the reaction mixture for 3 hours. This was attributed to the fact that the reaction mixture thickens to a paste and hence (-) carbinol gets trapped in the precipitation of the reaction mixture. However the quality of N-methyl paroxetine was not affected due to unreacted (-) carbinol upto 1%.
Holding of Reaction Mixture
To assess the impact of holding the reaction mixture for 48 hours, after complete addition of methane sulphonyl chloride, the reaction mass was kept under hold without stirring under nitrogen blanketing at 29°-35°C, and mesylate intermediate was isolated as per usual work up procedure. HPLC data revealed that there was no impact on quality of mesylate intermediate even after 48 hours of holding the reaction mass.
Recovery of Toluene: Recovery of Toluene was done at 40-45°C. To assess the impact of higher temperature on product quality recovery of toluene was also done at 50-55°C . It was observed that a higher recovery temperature did not affect the quality of the mesylate intermediate.
CHOICE OF SOLVENT FOR CONDENSATION AND SOLVENT RATIO
Initial experiments using non-polar solvents like toluene were not successful and when polar solvents [30] were tried followed by aqueous workup, a fine precipitate of the N-methyl paroxetine was obtained. The quality and yield also improved significantly. Table 3 summarizes the results of using various polar organic solvents.
Table 3: Selection of Proper Polar Solvent for Condensation Reaction
| Expt No. | Reaction Condition | HPLC Results | Comments |
|---|---|---|---|
| Expt No.9 | Dimethylsulphoxide (DMSO), KOH powder | 99.7% NMP | Yield: 89.1% |
| Expt No.10 | Methanol, KOH powder | 96.7% NMP | Yield: 67.3% |
| Expt No.11 | Sulpholane, KOH powder | 99.4% NMP | Yield: 90.7% |
| Expt No.12 | Dimethylacetamide (DMAC), KOH powder | 99.7% NMP | Yield: 87.7% |
| Expt No.13 | 2-Methoxy ethanol, KOH powder | 90.1% NMP | Yield: ~68.0% |
| Expt No.14 | N-Methyl Pyrrolidone KOH flakes | 98.99% NMP | Yield: 96.66% |
Since the prior art [14, 15]does not mention the use of polar solvents followed by aqueous work up and isolation of NMP as solid powder, the procedure described in this work is an entirely innovative strategy in the synthesis of N-methyl paroxetine.
The solvent ratio was fixed at 6 volumes with respect to (-) carbinol input as it was a minimum volume required to ensure that the reaction mixture could be stirred adequately. Lower volumes (4 volumes) did not affect the quality of NMP, but reaction mass was difficult to stir. Higher volumes ( 10 volumes) did not affect the quality of NMP. However, a larger amount of water is necessary to precipitate the product completely, indirectly increasing the effluents load.
HPLC data showed that the volume of DMSO used for the reaction was not critical. However, to ensure homogeneity and adequate stirring of the reaction mixture, 6 volumes of DSMO were selected.
Sesamol stoichiometry: During optimization experiments for the number of mole equivalents of sesamol used, it was observed that a minimum of 1.0 mole equivalent was necessary to obtain optimum yields and quality of NMP. Higher moles equivalents of sesamol did not have any impact on quality of N-methyl paroxetine Table 4.
Table 4: Deciding Sesamol Stoichiometry
| Expt No. | Volume of DMSO | Moles of Sesamol | HPLC Results | Comments |
|---|---|---|---|---|
| Expt No.15 | 6 volumes | 1.0 | 99.29% NMP 0.018% mesylate 0.22% UK RRT 1.32 | Yield: 94.7% |
| Expt No.16 | 6 volumes | 1.5 | 99.38%NMP, 0.018 %Sesamol, 0.23 UK RRT 1 .32 | Yield: 94.9% |
| Expt No.17 | 6 volumes | 1.1 | 99.47% NMP 0.18% UK RRT 1.04 0.26% UK RRT 1.27 | Yield: 95.26% |
| Expt No.18 | 6 volumes | 1.3 | 99.21% NMP 0.12% UK RRT 0.63 0.3% UK RRT 1.27 | Yield: 93.33% |
Sesamol forms a side chain in the API chemical structure and contributes significantly to the overall structure of molecule. Hence it is necessary from the regulatory [39, 40] point of view to control other phenolic impurities present in the sesamol raw material used. Phenolic impurities also react with the “ mesylate intermediate” thus forming impurities that can carry over to the final API. Therefore regulatory guidelines for selection of this raw material must be adhered to strictly.
Therefore,1.2 moles of sesamol was decided for the condensation reaction with the mesylate intermediate. Excess of sesamol was removed from the NMP as potassium salt, which is highly soluble in water.
Potassium hydroxide stoichiometry: Experimentaldata showed that the quality of N-methyl paroxetine was not affected even when 1.5 mole equivalents of KOH were used in DMSO. However, the yield was marginally less. Therefore, 2.2 moles of KOH were selected considering a 15% water content in the potassium hydroxide flakes.
Temperature of reaction: Experimental HPLC data showed that the quality of N-methyl paroxetine was affected at more than 65°C. Hence the reaction temperature was fixed at 50-55°C.
Quantity of water for quenching: HPLC data shows that the volume of water used for quenching the reaction is not critical. Hence an optimum quantity of 12 volumes with respect to (-) carbinol was selected to ensure complete precipitation of N-methyl paroxetine and removal of excess sesamol and KOH from the wet cake of N-methyl paroxetine.
Quantity of Water for Repulping Wet Cake of NMP:
The quantity of water used for repulping was not critical. Repulping was introduced in the process to remove alkalinity of the N-methyl paroxetine wet cake because pH of washings of the NMP cake remained on alkaline side in the range of pH 9 to pH11. Hence 15 volumes of water with respect to (-) carbinol was selected for repulping the wet cake of NMP.
Final Optimised Process:
As per the data obtained after optimisation experiments, following step wise final process for the manufacturing of N-methyl paroxetine was adopted for laboratory validation batches.
- In a clean 4-NRBF charge toluene (8 volumes with respect to input quantity of (-) carbinol, 1200 ml).
- Charge (-) Carbinol ,150 g( 0.671 moles). Stir to make a clear solution. Flush the reaction flask with Nitrogen.
- Charge Tri ethyl amine 206 ml (2.2 mole equivalent with respect to (-) carbinol, 1.47moles, density 0.727g/ml).
- Cool Reaction mixture to 0-10°C in crushed ice salt mixture.
- Mix methane sulphonyl chloride (1.3 mole equivalent with respect to (-) carbinol, 0.872moles), 70.5 ml i.e. 104 g. with (2 volumes with respect to (-) carbinol,) 300 ml Toluene in a dry & stoppered glass container.
- Transfer above methane sulphonyl chloride solution to addition funnel/vessel.
- Charge above methane sulphonyl chloride solution (370.5ml) to reaction mixture slowly.
- Maintain the temperature between 0-10°C during addition. Control the exotherm of reaction by halting the addition of methanesulphonyl chloride. It takes about 1-1.5 Hours for complete addition of reagent.
- Stir the reaction mixture between 0-10°C for about 2 hours.
- Withdraw RM-l Hr, 1 ml sample and submit for HPLC analysis.
- Withdraw RM-2 Hr, 1 ml sample and submit for HPLC analysis.
- If IPC complies (-) carbinol NMT 0.5% then Quench the reaction with ( 5 volumes with respect to (-) carbinol)750 ml water and stir for about 15 minutes.
- Separate the layers. Product is in Toluene Layer (Layer 1).
- Stir aqueous layer with toluene ( 1 volume with respect to (-) carbinol) 150 ml for about 10 minutes.
- Separate the layers. Discard aqueous layer. Combine Toluene layer (Layer 2) with above Layer 1.
- Stir aqueous layer with toluene ( 1 volume with respect to (-) carbinol)150 ml for about 10 minutes.
- Separate the layers. Discard aqueous layer. Combine Toluene layer ( Layer 3) with above Layers.
- Wash combined toluene layers (Layers 1+ 2 + 3) with water ( 1 volume with respect to (-) carbinol) 150 ml for about 10 minutes.
- Separate the layers. Discard aqueous layer.
- Wash combined toluene with distilled water ( l volume with respect to (-) carbinol ) 150 ml for about 10 minutes.
- Separate the layers. Discard aqueous layer.
- Recover Toluene on rotavapor ( Water bath temp. 45°C, vacuum 80-30 mbar) Toluene recovered: 1745m1. It takes about 1.5-2.0 hours for recovery of Toluene.
- Recover residual Toluene under high vacuum(Water bath temp. 45°C, vacuum 30- 10 mbar). It takes about 20-30 minutes to recover residual Toluene.
- Charge 150 ml DMSO ( 1 volume with respect to input of (-) carbinol) to oily concentrate of the “in situ” mesylate intermediate.
- Recover toluene under high vacuum( 30-10 mmHg) on rotavapor ( water bath temperature 45° C). It takes about 20-30 minutes to recover residual Toluene.
- Charge 150 ml DMSO. Use this solution of “ in situ “ mesylate intermediate for next step( volume ~ 450 ml).
Condensation
- 27. Charge DMSO 750 ml ( 5 volume with respect to input of (-) carbinol) into a clean 4NRBF ( capacity 5 lit .)
- 28. Charge sesamol ( 111 g, 0.803, 1.2 mole equivalents). Stir to make clear solution.
- 29. Heat reaction mixture in a water bath to 57° ±2°C( Water bath set temperature 57°C.). It takes about 15-20 minutes to attain 57° ±2°C temperature.
- 30. Charge Potassium hydroxide flakes( 110.5 g, 0.805moles,1.2 mole equivalents ).
- 31. Flush the reaction flask with N2 gas.
- 32. Charge above mesylate solution (volume ~ 450 ml )to addition funnel and add slowly into reaction mixture at 57° ±2°C. It takes about 15-20 minutes for addition.
- 33. Stir reaction mixture at 57° ±2°C for about 1 hour.
- 34. Withdraw RM- 1 Hr, 1 ml sample and submit for HPLC analysis.
- 35. Stir reaction mixture at 57° ±2°C for additional 30 minutes.
- 36. Withdraw RM-90 Minutes, 1 ml sample and submit for HPLC analysis.
- 37. If IPC complies ( IPC limit unreacted mesylate NMT 0.5%) then Cool RM to 40°C.
- 38. Quench RM with water (1800 ml) through addition funnel. Reaction mixture temperature goes upto 50°C due to heat of dilution of DMSO.
- 39. Cool RM to 30-35°C.
- 40. Filter the off white product.
- 41. Wash product cake with 1 % KOH solution ( 10 volumes with respect to (-) carbinol, 1500 ml solution , 500 ml x 3).
- 42. Suspend wet cake in distilled water ( 15 volumes with respect to (-) carbinol, 2250 m1). Stir at 30 -33°C for about 1 hour and filter/centrifuge.
- 43. Suck dry under vacuum for 30-45 minutes.
- 44. Transfer the product cake to SS Tray. Wet wt: 338 g.
- 45. Dry the product in vacuum oven at 55-60°C till moisture content of cake is NMT`0.5% w/w( vacuum 30-40 mmHg). It takes about 12-16 Hrs.
- 46. Weight after 12Hrs of drying under vacuum, Constant Dry wt: 200.4 g.
- 47. Yield : 200 g (87.0 % of theory from (-) carbinol ) (Expected :196.8 to 208.8g , 85 % to 90 % of theory).
- 48. Storage: At 30°C in double poly bags.
Lab Validation Analytical Results
Above process was scale up in Kilo lab and three consecutive experiments were conducted as laboratory scale processs validation experiments. Samples of N-methyl paroxetine were submitted for complete analysis and results are complied in following Table 5. Samples were subjected for lab stability studies at various temperature and humidity conditions.
Table 5: Comparative Analytical Data of Lab Validation Batches
| Specification | Limits | Expt No.Val1 | Expt No.Val2 | Expt No.Val3 |
|---|---|---|---|---|
| Appearance | Off white to buff powder | Buff powder | Buff powder | Buff powder |
| Identification by HPLC | Should comply | complies | complies | complies |
| Loss of drying | NMT 1.0% | 0.54% | 0.24% | 0.26% |
| HPLC Purity | NLT 99.0% | 99.73% | 99.47% | 99.49% |
| Related substances by HPLC 1. Specified identified a) carbinol b) mesylate c) Ether impurity RRT 1.04 2. Specified unidentified impurity a. Impurity at RRT 1.6 b. Impurity at RRT 1.8 3. Unspecified unidentified single largest impurity 4. Total impurities | NMT 0.15% NMT 0.1% NMT 0.15% NMT 0.15% NMT 0.15% NMT 0.1% NMT 0.5% | Below LOQ Below LOQ Below LOQ Below LOQ Below LOQ 0.06% 0.13% | Below LOQ Below LOQ Below LOQ Below LOQ Below LOQ 0.06% 0.15% | Below LOQ Below LOQ Below LOQ Below LOQ Below LOQ 0.07% 0.10% |
Applying QbD Principles to Process Development
If we rationalize how impurity profile of N-methyl paroxetine was improved?, then we understand advantage of DMSO solvent over that of sulpholane solvent. In this process development studies, it was observed that formation of mesylate intermediate at low temperature is critical process parameter. The quality of N-methyl paroxetine gets affected, if process parameters are not strictly adhere to. This step is also critical from the safety point of view and reaction exotherm must be controlled to avoid any run away situation during large scale manufacturing.
We would like to mention that use of mesylate intermediate is of concern to regulatory agencies, due to possibility of potential genotoxic impurity [40]. However, we could prove, by HPLC analytical methods that mesylate intermediate was absent in N-methyl paroxetine and final API and it was well below LOQ level.
Here we would like to emphasise that QbD principles were applied in this process development. Firstly we identified the exact route cause of impurity formation and then we applied knowledge of process chemisty to control/ stop the formation impurities. We have demonstrated that quality can be built in the process and “right first time” approach can be adopted to avoid failures and bitter surprises during scale up in manufacturing plant. Such type of strategy would also reduce overall cost of manufacturing an API and make a process the most competitive in current commercial environment. It is necessary that we are able to sustain the price pressure in generic market globally and remain in API market for long time doing business (First in, last out generic business strategy).
CONCLUSION
As per the results obtained after optimisation experiments for the condensation reaction, the final process for manufacturing N-methyl paroxetine using 6 volumes of DMSO solvent, 1.2 mole equivalents of sesamol, 2.2 mole equivalents of Potassium hydroxide flakes and maintaining the reaction temperature at 50-55°C. In addition, 12 volumes of water were needed for the quenching reaction and 15 volumes of water were used for repulping the wet cake of NMP was adopted for validation batches. The process was scaled up in Kilo lab and three consecutive experiments were conducted as laboratory scale processs validation experiments. Intermediate analysis results complied with the predetermined quality specifications.
ABBREVIATIONS
| API | = Active pharmaceutical ingredient |
| MsCl | = methane sulphonyl chloride |
| TEA | = Triethyl amine |
| NMP | = N-methyl paroxetine |
| HPLC | = High performance liquid chromatography |
| NMT | = Not more than |
| LOQ | = Limit of quantification |
| NLT | = Not less than |
| 4-NRBF | = 4 neck round bottom flask |
| UK | = Unknown impurity |
| RRT | = Relative retention time |
| RM | = reaction mixture |
| IPC | = In process Control |
| TLC | = Thin layer chromatography |
| QbD | = Quality by design |
| DMSO | = Dimethyl sulphoxide |
| ICH | = International conference on hormonisation |
| Expt. No. | = Experiment number |
| eq | = equivalent |
ACKNOWLEDGEMENT
The Authors are grateful to Principal, Ismail Yusuf College, Gogeshwari East, Mumbai for valuable guidance and constant encouragement during preparation of this article.
REFERENCES
| [1] | Ulla L, Jan S, Ralph H, Rinke S, Florian H, Werner B, Reinhard K. Lewis acid-catalyzed synthesis of 4-aminopyrimidines: a scalable industrial process, Org. Process Res. Dev 2013; 17(3): 427-431. |
| [2] | John, PG Jr; Edward, JD.; Michael, JH.; Erwin, I.; William, FK.; Albert, K.; Amy, DM. Understanding and control of dimethyl sulfate in a manufacturing process: kinetic modeling of a fischer esterification catalyzed by H2SO4, Org, Process Res. Dev 2012; 16(12): 232-239. |
| [3] | Robert B, David JD, Peter JD, Farhat H, Ying K, Clive M, John CM, Martin JS. l Identification of new catalysts to promote imidazolide couplings and optimisation of reaction conditions using kinetic modelling, Org. Process Res. Dev 2004; 8(6): 1054-1058. |
| [4] | Roy CF, Shiping X, Alan Ml. A catalyzed and highly selective ester reduction in the synthesis of an n-acylpyrrolidine: safe design through reaction calorimetry and modelling, Org. Process Res. Dev 2008; 12(6): 1307-1312. |
| [5] | Cole DC, Pagano N, Kelly MF, Ellingboe Jl. Use of design of experiments to optimize high-throughput semipreparative LC and LC/MS methods, J. Comb. Chem 2004; 6(1): 78-82. |
| [6] | Lydia BF, Milena C, Casimir CAl. Metal-catalyzed oxidation of a structured lipid model emulsion, J. Agric. Food Chem 2002; 50(24): 7114-7119. |
| [7] | Kae MB, Marke BM, Jennifer FTl. Optimization and scale-up of a suzuki-miyaura coupling reaction: development of an efficient palladium removal technique, Org. Process Res. Dev 2008; 12(5): 896-899. |
| [8] | (a) Mahesh SP, Nandita PS. Process considerations during API development, Pharmacuetical Technology Europe, ( ADVANSTAR COMMUNICATIONS) 2005; Feb pp: 19-27. (b) Montu P.B, Rajendra S. Design and development of a common generic product development strategy, Asian J. Pharm. Sci. Res2011; 1(1): 10-19. |
| [9] | Drugs of Future 1986; 11: 112-115. |
| [10] | (a) Amat M, Bosch J, Hidalgo J, Canto M, Perez M, Llor N, Molins E, Miravitlles Orozco, M. Luque J. Synthesis of Enantiopure trans-3,4-Disubstituted Piperidines. An Enantiodivergent Synthesis of (+)- and (-)-Paroxetine, J. Org. Chem 2000; 65: 3074. (b) Johnson TA, Curtis MD, Beak P. Highly diastereoselective and enantioselective carbon−carbon bond formations in conjugate additions of lithiated N-Boc allylamines to nitroalkenes: enantio-selective synthesis of 3,4- and 3,4,5-substituted piperidines including (−)-paroxetine, J. Am. Chem. Soc 2001; 123: 1004. |
| [11] | (a) Hughes G, Kimura M, Buchwald SL. Catalytic enantio-selective conjugate reduction of lactones and lactams, J. Am. Chem. Soc 2003;125: 11253. (b) Chen CY, Chang BR, Tsai MR, Chang MY, Chang NC. Regioselective reduction of N-alkyl-3-sulfonyl glutarimides to δ-lactams. Formal synthesis of (±)-paroxetine and (±)- tacamonine, Tetrahedron 2003; 59: 9383. |
| [12] | Cossy J, Mirguet O, Pardo DG, Desmurs JR. A formal synthesis of (-)-paroxetine by enantioselective ring enlargement of a trisubstituted prolinol, Eur. J. Org. Chem 2002: 3543. |
| [13] | Hynes PS, Stupple PA, Dixon DJ. Organocatalytic asymmetric total synthesis of (R)-rolipram and formal synthesis of (3S,4R)-paroxetine,Org. Lett 2008; 10: 1389. |
| [14] | Christensen JA, Squires RF. , . US 4007196 (A/S Ferrosan, 1977). 1977. |
| [15] | Christensen JA, Squires RF. , . US 3912743 (A/S Ferrosan, 1975). 1975. |
| [16] | Bower JF, Riis-Johannessen T, Szeto P, Whitehead AJ, Gallagher T. Stereospecific construction of substituted piperidines Synthesis of (-)-paroxetine and (+)-laccarin, Chem. Commun 2007: 728. |
| [17] | DeGonzalo G, Brieva R, Sanchez VM, Bayod M, Gotor V. Enzymatic resolution of trans-4-(4‘-fluorophenyl)-3-hydroxymethylpiperidines, key intermediates in the synthesis of (-)-paroxetine, J. Org. Chem 2001; 66: 8947. |
| [18] | Senda T, Ogasawara M, Hayashi T. Rhodium-catalyzed asymmetric 1,4-addition of organoboron reagents to 5,6-dihydro-2(1H)-pyridinones. Asymmetric Synthesis of 4-Aryl-2-piperidinones, J. Org. Chem 2001; 66: 6852. |
| [19] | Brandau S, Landa A, Franzén J, Marigo M, JØrgensen KA. organocatalytic conjugate addition of malonates to a,ß-unsaturated aldehydes: asymmetric formal synthesis of (-)-paroxetine, chiral lactams, and lactones, Angew. Chem. Int.Ed 2006; 45: 4305. |
| [20] | Koech PK, Krische MJ. Enantioselective total and formal syntheses of paroxetine (PAXIL) via phosphine-catalyzed enone a-arylation using arylbismuth(V) reagents: a regiochemical complement to Heck arylation, Tetrahedron 2006; 62: 10594. |
| [21] | Liu LT, Hong PC, Huang HL, Chen SF, Wang CLJ, Wen YS. Asymmetric syntheses of trans-3,4-disubstituted 2-piperidinones and piperidines, Tetrahedron Asymm 2001; 12: 419. |
| [22] | Greenhalgh DA, Simpkins NS. A novel chiral base mediated glutarimide desymmetrisation: application to the asymmetric synthesis of (-) -paroxetine, Synlett 2002: 2074. |
| [23] | Gill CD, Greenhalgh DA, Simpkins NS. Application of the chiral base desymmetrisation of imides to the synthesis of the alkaloid jamtine and the antidepressant paroxe-tine, Tetrahedron 2003; 59: 9213. |
| [24] | Czibula L, Nemes A, Sebök F, Szantay CJr, Mak M. A convenient synthesis of (-)-paroxetine, Eur. J. Org. Chem 2004; 15: 3336. |
| [25] | Takasu K, Nishida N, Tomimura A, Ihara M. Convenient synthesis of substituted piperidinones from a,ß-unsaturated amides formal synthesis of deplancheine, tacamonine, and paroxetine, J. Org. Chem 2005; 70: 3957. |
| [26] | Valero G, Schimer J, Cisarova I, Vesely J, Moyano A, Rios R. Highly enantioselective organocatalytic synthesis of piperidines Formal synthesis of (-)-Paroxetine, Tetrahedron Lett 2009; 50: 1943. |
| [27] | Yu MS, Lantos I, Peng ZQ, Yu J, Cacchio T. Asymmetric synthesis of (-)-paroxetine using PLE hydrolysis, Tetrahedron Lett 2000; 41: 5647. |
| [28] | Sudhir S, Ramesh Y. Synthesis and characterization of novel impurities in Paroxetine Hydrochloride hemihydrate and adopting QbD principles to built in quality in process of final drug substance, Asian J. Res. Chem 2012; 05(03): 329. |
| [29] | Ravi C. Paroxetine hydrochloride hemihydrates monograph, USP 29 NF 24 pp: 1644. |
| [30] | Impurities: Guidelines for Residual Solvents, Q3C, Recommended by the ICH on 17th July (1997), revised Table published by European Medicines Agency: Europe, 2012 February ; |
| [31] | William L. Roper, Toxicological profile for pyridine, Report by Agency for Toxic Substances and Disease Registry, U.S. Public Health Service: USA. 1992 |
| [32] | Piere S, Anne-Marie A, David SG. Tosylation/mesylation of 4-hydroxy-3-nitro-2-pyridinones as an activation step in the construction of dihydropyrido[3,4-b] benzo[f][1,4]thiazepin-1-one based anti-HIV agents, Tetrahedron Lett 2005; 46(16): 2919-2922. |
| [33] | Tipson RS. On esters of p-toluenesulfonic acid, J. Org. Chem 1944; 9: 235. |
| [34] | Majerski Z, Nikoletic M, Borçic S, Sunko DE. The“bicyclobutonium ion Reaction of (1-methylcyclo-propyl)carbinyl and 1-methyl cyclobutyl methanesulfonates with sodium borohydride under solvolytic conditions, Tetrahedron 1967; 23: 661. |
| [35] | Ronald KC, Kenneth LS. Facile synthesis of methanesulfonate esters, J. Org. Chem 1970; 35(9): 3195-3196. |
| [36] | Truce WE, Norell JR. Thietane dioxide derivatives via the interaction of sulfonyl chlorides with ketene diethylacetal, J. Am. Chem. Soc1963; 85: 3231. |
| [37] | King JF, Sun Lee TW. Mechanism of formation of sulfenes by dehydrohalogenation of alkanesulfonyl chlorides, J. Am. Chem. Soc 1969;91: 6524. |
| [38] | Opits G. Sulfines and sulfenes – the S-oxides and S,S-dioxides of thioaldehydes and thioketones, Angew. Chem. Ed. Engl 1967; 6: 107. |
| [39] | Tom C, Betsy F, Wendy M, Michael M, Dave R, Jean W. Starting material, reprocessing, retesting, and critical controls, Pharmaceutical Technology February. 2003 February;ADVANSTAR COMMUNICATIONS |
| [40] | FDA Perspectives: Designation of Regulatory Starting Materials in the Manufacturing of Drug Substances: Impact on ANDA Review Time, Barbara Scott, Pharmaceutical Technology. 2012 January;(ADVANSTAR COMMUNICATIONS) |
Developing a Commercially Viable Process for an Active Pharmaceutical Ingredient, Challenges, Myths and Reality in the Art of Process Chemistry
Ramesh Yamgar , Sudhir Sawant*
P. G. Department of Chemistry, Govt. of Maharashtra’s Ismail Yusuf College of Arts, Science and Commerce, Jogesh-wari (East), Mumbai 400 060, India
* Address correspondence to this author at the P. G. Department of Chemistry, Govt. of Maharashtra’s Ismail Yusuf College of Arts, Science and Commerce, Jogeshwari (East), Mumbai 400 060 India; Tel: +919325332034; Fax: +912228744755; E-mail: sawantsudhir@hotmail.com
PAXIL (paroxetine hydrochloride) is an orally administered psychotropic drug. It is the hydrochloride salt of a phenylpiperidine compound identified chemically as (-)-trans-4R-(4'fluorophenyl)-3S-[(3',4'-methylenedioxyphenoxy) methyl] piperidine hydrochloride hemihydrate and has the empirical formula of C19H20FNO3•HCl•½H2O. The molecular weight is 374.8 (329.4 as free base). The structural formula of paroxetine hydrochloride is:
 |
Paroxetine hydrochloride is an odorless, off-white powder, having a melting point range of 120° to 138°C and a solubility of 5.4 mg/mL in water.
Tablets
Each film-coated tablet contains paroxetine hydrochloride equivalent to paroxetine as follows: 10 mg–yellow (scored); 20 mg–pink (scored); 30 mg–blue, 40 mg–green. Inactive ingredients consist of dibasic calcium phosphate dihydrate, hypromellose, magnesium stearate, polyethylene glycols, polysorbate 80, sodium starch glycolate, titanium dioxide, and 1 or more of the following: D&C Red No. 30 aluminum lake, D&C Yellow No. 10 aluminum lake, FD&C Blue No. 2 aluminum lake, FD&C Yellow No. 6 aluminum lake.
Suspension For Oral Administration
Each 5 mL of orange-colored, orange-flavored liquid contains paroxetine hydrochloride equivalent to paroxetine, 10 mg. Inactive ingredients consist of polacrilin potassium, microcrystalline cellulose, propylene glycol, glycerin, sorbitol, methylparaben, propylparaben, sodium citrate dihydrate, citric acid anhydrous, sodium saccharin, flavorings, FD&C Yellow No. 6 aluminum lake, and simethicone emulsion, USP.


| Full chemical name | (3S,4R)-3-[(2H-1,3-benzodioxol-5-yloxy)methyl]-4-(4-fluorophenyl) piperidine.hydrochloride | |
| Formula | C19H20FNO3.HCl.0.5H2O | |
| Molecular weight | 374.83 | |
| Chemical abstract number | 110429-35-1 | |
| Supplied as | hydrochloride salt | |
| Appearance | white to off-white crystalline powder | |
| IR | corresponds | |
| NMR | corresponds to structure | |
| MS | MH+ corresponds | |
| UV max [nm] (mol, solvent) | 235.0 ± 1.0 (3800 ± 400, methanol) ; 294.0 ± 1.0 (4000 ± 400, methanol) | |
| Melting point[Celsius] | 143 ± 3 | |
| HPLC purity [%] | >98.5 | |
| Optical rotation (conc., solvent) | -90 ± 3(c=1.0, methanol) | |
| Free base content [%] | >84.2 | |
| Water content [%] | <5 | |
| Calculated hydrochloride content [%] | 9.7 | |





//////// API Process development, process optimisation, negative experiments, critical process parameters, carryover of impurities, reagent stoichiometry.

























 LIONEL MY SON
LIONEL MY SON