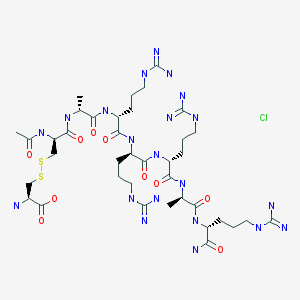
IPI 926, Saridegib, Patidegib
C29H48N2O3S
Exact Mass: 504.33856
1037210-93-7
- Patidegib hydrochloride
- Saridegib hydrochloride
- C29-H48-N2-O3-S.Cl-H
- 541.2361
Methanesulfonamide,
N-((2S,3R,3'R,3aS,4'aR,6S,6'aR,6'bS,7aR,12'aS,12'bS)-2',3',3a,4,4',4'a,5,5',6,6',6'a,6'b,7,7',7a,8',10',12',12'a,12'b-eicosahydro-3,6,11',12'b-tetramethylspiro(furo(3,2-b)pyridine-2(3H),9'(1'H)-naphth(2,1-a)azulen)-3'-yl)-,
hydrochloride (1:1)
CAS 1169829-40-6 HCL
Saridegib also known as IPI-926
is an experimental drug candidate undergoing clinical trials for the
treatment of various types of cancer, including hard to treat
hematologic malignancies such as myelofibrosis and ligand-dependant
tumors such as chondrosarcoma.[1] IPI-926 exhibits its pharmacological effect by inhibition of the G protein-coupled receptor smoothened, a component of the hedgehog signaling pathway.[2]Chemically, it is a semi-synthetic derivative of the alkaloid cyclopamine. The process begins with cyclopamine extracted from harvested Veratrum californicum which is taken through a series of alterations resulting in an analogue of the natural product cyclopamine, making IPI-926 the only compound in development/testing that is not fully synthetic.[2]

Saridegib is a member of a class of anti-cancer compounds known as hedgehog inhibitors (Hhi). Most of these compounds affect thehedgehog signaling pathway via inhibition of smoothened (Smo), a key component of the pathway. Depending on when a Hh inhibiting compound is approved by the U.S. Food and Drug Administration
(FDA), there may be a perceived need for one to be differentiated over
another for marketing purposes, which could lead to different
nomenclature (e.g., a Hhi or an agonist of Smo).
This marketing technique is more of a differentiation strategy than a scientific property of these compounds, as the mechanism of action (MOA) in the end is inhibition of the Hh pathway, targeting cancer stem cells. However, as these new compounds are further studied, identification of differences in a compound's MOA, could lead to hypotheses regarding the stage at which Smo is inhibited, where along the pathway the compound binds, or specific binding properties of a compound.
If these hypotheses are proven, claims could be made regarding a specific compound's MOA and how it affects efficacy, safety, combinability with other cancer treatments, etc. Scientific data in support of such hypotheses have not been published to date.
SARIDEGIBThis marketing technique is more of a differentiation strategy than a scientific property of these compounds, as the mechanism of action (MOA) in the end is inhibition of the Hh pathway, targeting cancer stem cells. However, as these new compounds are further studied, identification of differences in a compound's MOA, could lead to hypotheses regarding the stage at which Smo is inhibited, where along the pathway the compound binds, or specific binding properties of a compound.
If these hypotheses are proven, claims could be made regarding a specific compound's MOA and how it affects efficacy, safety, combinability with other cancer treatments, etc. Scientific data in support of such hypotheses have not been published to date.
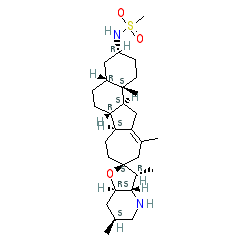
N-[(3R,3'R,3'aS,4aR,6'S,6aR,6bS,7'aR,9S,12aS,12bS)-3',6',11,12b-tetramethylspiro[1,2,3,4,4a,5,6,6a,6b,7,8,10,12,12a-tetradecahydronaphtho[2,1-a]azulene-9,2'-3a,4,5,6,7,7a-hexahydro-3H-furo[3,2-b]pyridine]-3-yl]methanesulfonamide
There
are currently no drugs in the Hhi class FDA approved, however IPI-926
and GDC-0449 are the 2 leading compounds in the class. IPI-926,
GDC-0449, and LDE-225 are the only compounds that have generic names
passed by the United States Adopted Name
(USAN) council (Infinity IPI-926/saridegib, Genentech
GDC-0449/vismodegib, and Novartis LDE-225/erismodegib). Although
Infinity is further along in chondrosarcoma, myelofibrosis, and AML,
Roche/Genentech recently submitted an NDA for GDC-0449 for the treatment
of adults with advanced basal cell carcinoma (BCC) when surgery is no
longer an option, and the FDA has accepted and has filed the NDA, giving
it priority review status. Thus it appears that Roche/Genentech will be
the first Hhi to market with GDC-0449, if approved, for the treatment
of advanced BCC, with Infinity second to market with IPI-926 for
treatment in chondrosarcoma. It appears Infinity will not pursue an
indication for BCC and focus on cancers with high unmet needs.[1][3][4][5][6]
Other Hhi-class compounds not as far along in development as IPI-926 and GDC-0449 include:[7]

The hedgehog pathway inhibitor IPI-926 has
been in clinical investigation for basal cell carcinoma, chondrosarcoma,
and pancreatic cancer. In the final step of the synthesis of IPI-926
the drug substance (DS) is isolated as the hydrochloride salt of the
2-propanol (2-PrOH) solvateOther Hhi-class compounds not as far along in development as IPI-926 and GDC-0449 include:[7]
- Novartis' LDE-225 (USAN generic name erismodegib)
- Exelixis/Bristol-Myers Squibb's BMS-833923 (XL139)
- Millennium Pharmaceuticals's TAK-441
- Pfizer's PF-04449913
Fig 1. Chemical structure comparison between IPI-926 and cyclopamine
IPI-926
is currently developed by Infinity Pharmaceuticals, Inc. Malignant
activation of the Hedgehog pathway is implicated in multiple cancer
settings and Infinity's development strategy is designed to enable
IPI-926 to target a broad range of critical oncology targets - from the
tumor cell to the cancer microenvironment. This broadly applicable,
targeted approach represents an innovative method for fighting cancer
and has potential in treating a range of cancers, including pancreatic
cancer, small cell lung cancer, ovarian cancer, bladder cancer,
medulloblastoma, basal cell carcinoma, and certain hematological
malignancies.

A
design of experiments (DoE) approach was taken to optimize purity and
reaction yield of the final debenzylation and hydrochloride salt
formation of IPI-926. The study involved a careful dissection of the
different process steps to enable an independent investigation of these
steps while ensuring that process streams were representative. The
results enabled a streamlined process from the final chemical
transformation to the salting and isolation and led to the elimination
of variability in the process as well as a robust control of impurities.
The optimized process was applied to production and demonstrated on the
kilogram scale.
A Design of Experiments Approach to a Robust Final Deprotection and Reactive Crystallization of IPI-926, A Novel Hedgehog Pathway Inhibitor
Infinity Pharmaceuticals, 784 Memorial Drive, Cambridge, Massachusetts 02139, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00214
1H NMR (400 MHz, CDCl3) 6.90 (br s, 1H), 3.31 (dt, J = 10.6, 3.8 Hz, 1H), 3.20 (br s, 1H), 3.10 (dd, J = 13.7, 4.5 Hz, 1H), 2.91 (s, 3H), 2.62 (dd,J = 9.9, 7.6 Hz, 1H), 2.33 (br d, J = 14.5 Hz, 1H), 2.27–2.15 (m, 1H), 2.10 (dd, J = 14.5, 6.9 Hz, 1H), 1.99–1.17 (m, 28H), 1.05 (q, J = 11.6 Hz, 1H), 0.93 (d, J = 7.4 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H), 0.86 (s, 3H) ppm.
13C NMR (100 MHz, CDCl3) 140.47, 124.53, 82.48, 76.97, 63.73, 54.08, 53.87, 50.12, 49.98, 47.19, 44.73, 42.27, 42.10, 40.24, 37.55, 37.44, 36.04, 34.44, 31.87, 31.33, 30.46, 29.79, 28.37, 27.94, 26.26, 24.19, 22.70, 18.92, 10.19 ppm;
MS: m/z = 505.29 [M + H]+.
PAPER
Tremblay, M. R.; Lescarbeau, A.; Grogan, M. J.; Tan, E.; Lin, G.; Austad, B. C.; Yu, L.-C.;Behnke, M. L.; Nair, S. J.; Hagel, M.; White, K.; Conley, J.; Manna, J. D.; Alvarez-Diez, T. M.; Hoyt, J.; Woodward, C. N.; Sydor, J. R.; Pink, M.; MacDougall, J.; Campbell, M. J.;Cushing, J.; Ferguson, J.; Curtis, M. S.; McGovern, K.; Read, M. A.; Palombella, V. J.;Adams, J.; Castro, A. C. J. Med. Chem. 2009, 52, 4400– 4418, DOI: 10.1021/jm900305z
J. Med. Chem., 2009, 52 (14), pp 4400–4418
DOI: 10.1021/jm900305z

Recent
evidence suggests that blocking aberrant hedgehog pathway signaling may
be a promising therapeutic strategy for the treatment of several types
of cancer. Cyclopamine, a plant Veratrum alkaloid, is a natural
product antagonist of the hedgehog pathway. In a previous report, a
seven-membered D-ring semisynthetic analogue of cyclopamine, IPI-269609 (2),
was shown to have greater acid stability and better aqueous solubility
compared to cyclopamine. Further modifications of the A-ring system
generated three series of analogues with improved potency and/or
solubility. Lead compounds from each series were characterized in vitro
and evaluated in vivo for biological activity and pharmacokinetic
properties. These studies led to the discovery of IPI-926 (compound 28),
a novel semisynthetic cyclopamine analogue with substantially improved
pharmaceutical properties and potency and a favorable pharmacokinetic
profile relative to cyclopamine and compound2. As a result,
complete tumor regression was observed in a Hh-dependent medulloblastoma
allograft model after daily oral administration of 40 mg/kg of compound
28.
28 (4.06 g, 8.05 mmol, 95% for two steps). NMR δH (400 MHz, CDCl3) 6.90 (br s, 1H), 3.31 (dt, J = 10.6, 3.8 Hz, 1H), 3.20 (br s, 1H), 3.10 (dd, J = 13.7, 4.5 Hz, 1H), 2.91 (s, 3H), 2.62 (dd, J = 9.9, 7.6 Hz, 1H), 2.33 (br d, J = 14.5 Hz, 1H), 2.27−2.15 (m, 1H), 2.10 (dd, J = 14.5, 6.9 Hz, 1H), 1.99−1.17 (m, 28H), 1.05 (q, J = 11.6 Hz, 1H), 0.93 (d, J = 7.4 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H), 0.86 (s, 3H); NMR δC (100 MHz, CDCl3)
140.47, 124.53, 82.48, 76.97, 63.73, 54.08, 53.87, 50.12, 49.98, 47.19,
44.73, 42.27, 42.10, 40.24, 37.55, 37.44, 36.04, 34.44, 31.87, 31.33,
30.46, 29.79, 28.37, 27.94, 26.26, 24.19, 22.70, 18.92, 10.19; m/z = 505.29 [M + H]+; HPLC 99.1 a/a % at 215 nm.



Click on images for clear view.................
Paper
A
design of experiments (DoE) approach was taken to optimize purity and
reaction yield of the final debenzylation and hydrochloride salt
formation of IPI-926. The study involved a careful dissection of the
different process steps to enable an independent investigation of these
steps while ensuring that process streams were representative. The
results enabled a streamlined process from the final chemical
transformation to the salting and isolation and led to the elimination
of variability in the process as well as a robust control of impurities.
The optimized process was applied to production and demonstrated on the
kilogram scale.
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00214..........http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00214
IPI-926 free base:
1H NMR (400 MHz, CDCl3) 6.90 (br s, 1H), 3.31 (dt, J = 10.6, 3.8 Hz, 1H), 3.20 (br s, 1H), 3.10 (dd, J = 13.7, 4.5 Hz, 1H), 2.91 (s, 3H), 2.62 (dd,J = 9.9, 7.6 Hz, 1H), 2.33 (br d, J = 14.5 Hz, 1H), 2.27–2.15 (m, 1H), 2.10 (dd, J = 14.5, 6.9 Hz, 1H), 1.99–1.17 (m, 28H), 1.05 (q, J = 11.6 Hz, 1H), 0.93 (d, J = 7.4 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H), 0.86 (s, 3H) ppm.
13C NMR (100 MHz, CDCl3)
140.47, 124.53, 82.48, 76.97, 63.73, 54.08, 53.87, 50.12, 49.98, 47.19,
44.73, 42.27, 42.10, 40.24, 37.55, 37.44, 36.04, 34.44, 31.87, 31.33,
30.46, 29.79, 28.37, 27.94, 26.26, 24.19, 22.70, 18.92, 10.19 ppm;
MS: m/z = 505.29 [M + H]+.
References
- "Pipeline: IPI-926". Infinity Pharmaceuticals.
- Tremblay, MR; Lescarbeau, A; Grogan, MJ; Tan, E; Lin, G; Austad, BC; Yu, LC; Behnke, ML et al. (2009). "Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926)". Journal of Medical Chemistry 52 (14): 4400–18. doi:10.1021/jm900305z. PMID 19522463.
- "Pipeline". Infinity Pharmaceuticals.
- "Genentech Pipeline". Genentech.
- "USAN Stem List" (PDF). AMA.
- "Names under consideration". AMA.
- "Search results for Hh clinical trials". United National Institute of Health's ClinicalTrials.gov.
- 1. Tremblay MR, Lescarbeau A, Grogan MJ, Tan E, Lin G, Austad BC, Yu LC, Behnke ML, Nair SJ, Hagel M et al.. (2009)
Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926).
J. Med. Chem., 52 (14): 4400-18.
 | |
 | |
| Names | |
|---|---|
| IUPAC name
N-((2S,3R,3aS,3′R,4a′R,6S,6a′R,6b′S,7aR,12a&prmie;S,12b′S)-3,6,11′,12b′-tetramethyl-2′,3a,3′,4,4′,4a′,5,5&prmie;,6,6′,6a′,6b′,7,7a,7′,8′,10′,12′,12a′,12b′-icosahydro-1′H,3H-spiro[furo[3,2-b]pyridine-2,9'-naphtho[2,1-a]azulen]-3'-yl)methanesulfonamide
| |
| Other names
saridegib
| |
| Identifiers | |
| 1037210-93-7 | |
| ChEMBL | ChEMBL538867 |
| ChemSpider | 26353073 |
| 8198 | |
| Jmol-3D images | Image |
| PubChem | 25027363 |
| UNII | JT96FPU35X |
| Properties | |
| C29H48N2O3S | |
| Molar mass | 504.77 g·mol−1 |
| Pharmacology | |
| Legal status |
|





 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPD