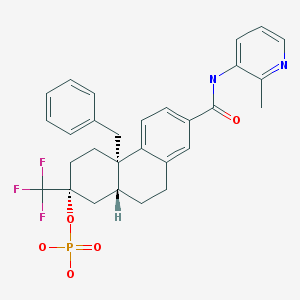
Fosdagrocorat, PF-04171327,
CAS 1044535-58-1
(2R,4aS,10aR)-4a-Benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl dihydrogen phosphate
2-Phenanthrenecarboxamide, 4b,5,6,7,8,8a,9,10-octahydro-N-(2-methyl-3-pyridinyl)-4b-(phenylmethyl)-7-(phosphonooxy)-7-(trifluoromethyl)-, (4bS,7R,8aR)-
(2R,4aS,10aR)-4a-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl dihydrogen phosphate
MF C29H30F3N2O5P
Exact Mass: 574.1844
Exact Mass: 574.1844
- PF 04171327
- PF-04171327
- UNII-HPI19004QS
- Selective Glucocorticoid Receptor Modulator
phase 2 .Rheumatoid Arthritis
Glucocorticoid receptor modulators
Pfizer
- 03 Sep 2015Phase II development of fosdagrocorat is ongoing
- 01 Jun 2014Pfizer completes a phase II trial in Rheumatoid arthritis in US, Bulgaria, Colombia, the Czech Republic, Germany, Hungary, India, South Korea, Malaysia, Mexico, Poland, Romania, Russia, Serbia, Slovakia, South Africa, Spain and the Ukraine (NCT01393639)
- 30 Sep 2011Phase-II clinical trials in Rheumatoid arthritis in Bulgaria, Colombia, Germany, India, Malaysia, Mexico, Poland, Romania and South Africa (PO)
Fosdagrocorat, also known as PF-04171327, a dissociated agonist of the glucocorticoid receptor (DAGR), a selective high-affinity partial agonist of the GR with potent anti-inflammatory activity at exposures that provide less undesirable effects on bone and glucose metabolism compared with prednisone (pred).
Glucocorticoid receptor modulators are glucocorticoid receptor ligands that are used to treat a variety of conditions because of their powerful anti-inflammatory, antiproliferative and immunomodulatory activity. J. Miner, et al., Expert Opin. Investig. Drugs (2005) 14(12):1527-1545.
Examples of glucocorticoid receptor modulators include dexamethasone, prednisone, prednisolone, RU-486, and as described in WO 2000/66522 and WO 2004/005229.
Treatment with glucocorticoid receptor modulators is often associated with side effects, such as bone loss and osteoporosis.
Identifying a glucocorticoid receptor modulator that is efficacious, potent, and has mitigated side-effects fulfills a medical need.
Examples of glucocorticoid receptor modulators include dexamethasone, prednisone, prednisolone, RU-486, and as described in WO 2000/66522 and WO 2004/005229.
Treatment with glucocorticoid receptor modulators is often associated with side effects, such as bone loss and osteoporosis.
Identifying a glucocorticoid receptor modulator that is efficacious, potent, and has mitigated side-effects fulfills a medical need.
SYNTHESIS COMING............
PATENT
WO 2008093227/US 20100286214
SCHEME A

The 1 (/?)-Benzyl-5-bromo-9(S)-hydro-10(R)-hydroxy-10(R)-methyl-tricyclo[7.3.1.02'7]trideca-2,4,6-trien-13-one of Formula A-8 was prepared using the protocol described in Scheme A, which is generally disclosed in WO 00/66522. Ph depicts Phenyl. Bn depicts Benzyl. Compound A-1 can be purchased (for example, VOUS and Riverside; CAS No. 4133-35-1 ). Compound A-2 can be prepared as described in Org. Syn. 1971 , 51 , 109-112.
SCHEME B


The (4βS,7R,8αR)-4β-benzyl-7-hydroxy-Λ/-(2-methylpyridin-3-yl)-7-(trifluoromethyl)-4b,5,6,7,8α,9,10-octahydrophenanthrene-2-carboxamide was prepared as described in Scheme B.
SCHEME C
 The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme C. Bn depicts benzyl.
The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme C. Bn depicts benzyl.
SCHEME D


The (2R,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme D. Bn depicts benzyl. Ph depicts phenyl.
SCHEME E


The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoy[)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme E. Bn depicts benzyl. Ph depicts phenyl.
Starting Material A-8 is 1(R)~Benzyl-5-bromo-9(S)-hydro-10(R)-hydroxy-10(R)-methyl-tricyclo[7.3.1.02'7]trideca-2,4,6-trien-13-one as depicted by the following formula:

Preparation 1 : (S)-4a-benzyl-7-bromo-2-ethoxy-3,4,4a,9-tetrahydrophenanthrene

Starting Material A-8 (450 g; 1.17 moles) was dissolved in ethanol (4.5 L) at ambient temperature. 21% sodium ethoxide in ethanol (44 mL; 0.12 moles) was added and the mixture was heated to reflux for three hours. Once the Starting Material A-8 was consumed, the reaction mixture was chilled to -250C. Acetyl chloride (250 mL; 3.51 moles) was slowly added to the mixture while the temperature was maintained near -25°C. After the addition was complete, the mixture was warmed to O0C and held there until the intermediate enone was consumed. The mixture was slurry at this point. 21 % sodium ethoxide in ethanol (1.31 L; 3.51 moles) was added to the mixture while the temperature was maintained between -5°C and 50C. If the mixture was not basic, more sodium ethoxide was added. The temperature of the mixture was increased to 25°C and then diluted with water (5.9 L). The mixture was filtered and the solid was washed with water (3 X). The title compound (440 g; 85 area %) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.27 (t, 3H), 1.65 (dt, 1 H), 2.06 (d, 1 H), 2.21 (dd, 1 H)1 2.49 (m, 1 H), 2.65 (m, 2H), 2.89 (m, 2H), 3.85 (q, 2H), 5.45 (m, 2H), 6.44 (d, 2H), 6.98 (t, 2H), 7.06 (m, 2H), 7.25 (d, 1 H), 7.33 (dd, 1 H).
Preparation 2: (S)-4a-benzyl-7-bromo-2,2-(1,2-ethylenedioxy)-1,2,3,4,4a,9-hexahydrophenanthrene

The (S)-4α-benzyl-7-bromo-2-ethoxy-3,4,4α,9-tetrahydrophenanthrene (1270 g; 3.2 moles; 85 area %, which may be prepared as described in Preparation 1 ) was dissolved in toluene (6.45 L). The ethylene glycol (898 mL; 16.1 moles) and p-toluenesulfonic acid (6.1 g; 0.03 moles) were added and the reaction heated to reflux. Solvent (1 L) was distilled from the mixture and replaced with fresh toluene (1 L). This distillation process was repeated twice more. More p-toluenesulfonic acid (6.1 g) was added each time fresh toluene was added. During the reaction, two intermediates (detected by LC) were formed as the substrate was converted into product. The end point of the reaction was an equilibrium point between the two intermediates and the product. Once the endpoint was reached, the mixture was cooled to ambient temperature. The mixture was washed with 0.5 M NaOH (2 L). The phases separated quickly and both were dark with a small rag layer. The mixture was washed with water (2 L). The phases
separated very slowly. The mixture was dried by azeotropic distillation. Methanol (4 L) was added to the mixture and solvent (4 L) was distilled from the mixture. The methanol addition and solvent distillation were repeated twice more. Methanol was added to the mixture and precipitation occurred a few minutes later. More methanol (4 L) was added to the mixture and then brought to reflux. After 30 minutes, the mixture was cooled to 00C. The mixture was filtered and the solid was washed with chilled methanol (2 X 2L). The solid was dried in a vacuum oven at 65°C. The title compound (882 g; 98 area %) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.71 (m, 2H), 2.06 (m, 2H), 2.31 (dd, 1 H), 2.39 (m, 1 H), 2.68 (d, 1 H), 2.77 (m, 1 H), 2.86 (dd, 1 H), 3.36 (d, 1 H), 3.86 (m, 4H), 5.45 (m, 1 H), 6.50 (m, 2H), 7.00 (m, 4H), 7.37 (dd, 1 H), 7.44 (d, 1 H).
separated very slowly. The mixture was dried by azeotropic distillation. Methanol (4 L) was added to the mixture and solvent (4 L) was distilled from the mixture. The methanol addition and solvent distillation were repeated twice more. Methanol was added to the mixture and precipitation occurred a few minutes later. More methanol (4 L) was added to the mixture and then brought to reflux. After 30 minutes, the mixture was cooled to 00C. The mixture was filtered and the solid was washed with chilled methanol (2 X 2L). The solid was dried in a vacuum oven at 65°C. The title compound (882 g; 98 area %) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.71 (m, 2H), 2.06 (m, 2H), 2.31 (dd, 1 H), 2.39 (m, 1 H), 2.68 (d, 1 H), 2.77 (m, 1 H), 2.86 (dd, 1 H), 3.36 (d, 1 H), 3.86 (m, 4H), 5.45 (m, 1 H), 6.50 (m, 2H), 7.00 (m, 4H), 7.37 (dd, 1 H), 7.44 (d, 1 H).
Preparation 3: (S)-methyl 4β-benzyl-7,7-(1,2-ethylenedioxy)-4β,5,6,7,8,10-hexahydrophenanthrene-2-carboxylate

The (S)-4α-benzyl-7-bromo-2,2-(1 ,2-ethylenedioxy)-1 ,2,3,4,4α,9-hexahydrophenanthrene (719 g; 1.75 moles, which may be prepared as described in Preparation 2) was dissolved in tetrahydrofuran (7.19 L) and chilled to -7O0C. The 1.6 M n-butyl lithium in hexane (2270 mL; 2.27 moles) was added at a rate such that the temperature was maintained below -6O0C. The mixture held an additional 15 minutes after the addition. Carbon dioxide (108 g; 2.45 moles) was added while the temperature was maintained below -60°C. The mixture held an additional 15 minutes after the addition. The mixture was warmed to ambient temperature. Solvent (7 L) was distilled from the mixture at atmospheric pressure. DMF (7 L) was added to the mixture. The mixture was cooled to ambient temperature. Methyl iodide (152 mL; 2.45 moles) was added and the mixture was held until the reaction was completed (~1 hour). The mixture was heated to 7O0C and solvent was distilled by gradually reducing the pressure to 70 mmHg. Once distillation had ceased, the mixture was cooled to room
temperature. Water (6.5 L) was slowly added to the mixture to precipitate the product. The mixture was filtered and the solid washed with water (3 X). The solid was dried on the filter. The crude product (736 g; 74 area %) was obtained as a beige solid. The product was purified by chromatography. 463 g of product was recovered from the chromatography. This material was separated from n-heptane (6130 mL). 394 g of the title compound was recovered. Another 70 g of title compound was recovered from the mother liquor by chromatography. 1H NMR (DMSO) δ ppm: 1.74 (m, 2H), 2.10 (m, 2H)1 2.33 (dd, 1 H), 2.45 (m, 1 H), 2.72 (d, 1 H), 2.79 (m, 1 H), 2.94 (dd, 1 H), 3.40 (d, 1 H), 3.87 (m, 7H), 5.49 (m, 1 H), 6.47 (m, 2H), 6.93 (m, 2H), 7.01 (m, 1 H), 7.42 (d, 1 H), 7.64 (d, 1 H), 7.79 (dd, 1 H).
temperature. Water (6.5 L) was slowly added to the mixture to precipitate the product. The mixture was filtered and the solid washed with water (3 X). The solid was dried on the filter. The crude product (736 g; 74 area %) was obtained as a beige solid. The product was purified by chromatography. 463 g of product was recovered from the chromatography. This material was separated from n-heptane (6130 mL). 394 g of the title compound was recovered. Another 70 g of title compound was recovered from the mother liquor by chromatography. 1H NMR (DMSO) δ ppm: 1.74 (m, 2H), 2.10 (m, 2H)1 2.33 (dd, 1 H), 2.45 (m, 1 H), 2.72 (d, 1 H), 2.79 (m, 1 H), 2.94 (dd, 1 H), 3.40 (d, 1 H), 3.87 (m, 7H), 5.49 (m, 1 H), 6.47 (m, 2H), 6.93 (m, 2H), 7.01 (m, 1 H), 7.42 (d, 1 H), 7.64 (d, 1 H), 7.79 (dd, 1 H).
Preparation 4: (4βS,8α/?)-methyl 4β-benzyl-7,7-(1,2-ethylenedioxy)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (S)-methyl 4β-benzyl-7,7-(1 ,2-ethylenedioxy)-4β,5,6,7,8,10-hexahydrophenanthrene-2-carboxylate (201 g; 0.515 moles, which may be prepared as described in Preparation 3) and 50 ml of ethylene glycol was dissolved in toluene (2.0 L) in an autoclave. To this was added 10 grams of a 5% Pd/C (dry catalyst). The autoclave was then sealed and purged with nitrogen (three cycles) followed by hydrogen (three cycles). The reaction was run for 18 hours with a pressure of 80 psig and temperature of 50 0C. HPLC analysis for completion and selectivity (typical selectivity's are: 95 to 5, Trans to Cis). The suspension was filtered through Celite® to remove the catalyst and the toluene solution is concentrated at 50 0C, under vacuum, to
approximately 200 ml. While still at 50 0C, 1 L of 1-butanol was added and the solution heated to 60 0C, until clear. Upon cooling, the resulting solid title compound was isolated by vacuum filtration (196 grams; 97%; Trans to Cis 95.75 to 4.24). 1H NMR (300 MHz, CDCI3) δ ppm: 7.79 (bs, 1 H1 Ar-H), 7.47 (d, J= 9 Hz, 1 H, Ar-H), 7.13-7.05 (cm, 3H, Ar-H), 6.56-6.53 (cm, 2H, Ar-H), 6.43 (d, J= 9 Hz, 1 H, Ar-H), 4.04-3.93 (cm, 4H, 2-CH2), 3.89 (s, 3H, CH3),3.08-3.03 (cm, 3H, CH2, CH-H), 2.63 (d, J= 15 Hz, CH-H), 2.22-1.72 (cm, 8H, 4-CH2), 1.57 (cm, 1 H, CH-H).; 13CNMR (CDCI3, δ): 167.7, 149.2, 137.7, 136.4, 131.1 , 130.5, 127.8, 127.7, 127.4, 126.3, 125.5, 108.9, 64.6, 64.5, 52.1 , 40.5, 39.8, 38.3, 35.8, 31.6, 30.3, 27.9, 24.6.
approximately 200 ml. While still at 50 0C, 1 L of 1-butanol was added and the solution heated to 60 0C, until clear. Upon cooling, the resulting solid title compound was isolated by vacuum filtration (196 grams; 97%; Trans to Cis 95.75 to 4.24). 1H NMR (300 MHz, CDCI3) δ ppm: 7.79 (bs, 1 H1 Ar-H), 7.47 (d, J= 9 Hz, 1 H, Ar-H), 7.13-7.05 (cm, 3H, Ar-H), 6.56-6.53 (cm, 2H, Ar-H), 6.43 (d, J= 9 Hz, 1 H, Ar-H), 4.04-3.93 (cm, 4H, 2-CH2), 3.89 (s, 3H, CH3),3.08-3.03 (cm, 3H, CH2, CH-H), 2.63 (d, J= 15 Hz, CH-H), 2.22-1.72 (cm, 8H, 4-CH2), 1.57 (cm, 1 H, CH-H).; 13CNMR (CDCI3, δ): 167.7, 149.2, 137.7, 136.4, 131.1 , 130.5, 127.8, 127.7, 127.4, 126.3, 125.5, 108.9, 64.6, 64.5, 52.1 , 40.5, 39.8, 38.3, 35.8, 31.6, 30.3, 27.9, 24.6.
Preparation 5: (4βS,8α/?)-methyl 4β-benzyl-7-oxo-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

ThΘ (4βS,8αR)-mΘthyl 4β-benzyl-7,7-(1 ,2-ethylenΘdioxy)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (150 g, 382 mmol, which may be prepared as described in Preparation 4) was dissolved in dichloromethane (630 ml). Water (270 ml) was added with stirring followed by trifluoroacetic acid (73 ml. 1150 mmol) via drop funnel over 30 minutes, maintaining the internal temperature below 3O0C. After the addition was complete, the reaction was heated at 4O0C for 2 hours. In process check indicated incomplete reaction with around 9% (area percent) starting material. The layers were separated and fresh water (270 ml) and trifluoroacetic acid (31 ml) was added. The reaction mixture was heated at 4O0C for 1 hour. This process was continued until the starting material was consumed. The organic phase was washed with 5% aqueous sodium bicarbonate (300 ml), water (300 ml) and dried over MgSO4 and concentrated to dryness to give 126.4 g of the title compound (representing a 95% yield). 1H NMR (DMSO) δ ppm: 7.70 (s, 1 H), 7.37 (d, J=8.4 Hz, 1 H), 7.11 (m, 3H), 6.6 (d, J= 5.70 Hz, 2H), 6.45 (d, J=8.4 Hz, 1H), 3.80 (s, 3H), 3.80 (m, 2H), 3.04-1.48 (m, 11 H).
Preparation 6: (4βS,7f?,8α/?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5J6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (4βS,8αf?)-methyl 4β-benzyl-7-oxo-4β,5,6,7,8I8α,9,10-octahydrophenanthrene-2-carboxylate (118g, 0.339 mole, which may be prepared as described in Preparation 5) dissolved in dichloromethane was chilled to -5O0C. The solution became turbid. 1.0 M Tetrabutylammonium fluoride a solution in THF (3.4 ml, 0.003 mol) was added with no appreciable temperature change. Trifluorotrimethylsilane (79 ml, 0.51 mol) was added over 20 minutes with a color change to bright orange to light red in color. The reaction mixture was held at -50 0C for about 2 hours and then allowed to warm to 0 0C.
Tetrabutylammonium fluoride (340 ml, 0.34 moles) was added very slowly at 0 0C, to the reaction mixture over 45 minutes. An exotherm was observed with gas evolution. The reaction mixture was stirred 10 minutes and HPLC analysis indicated complete desilylialation. Water (1 L) was added to the reaction mixture and with vigorous stirring and allowed to warm to room temperature. The organic layer was washed with water (1 L). The organic layer was concentrated and chromatographed to produce 72 g, 51 % of the title compound, with an additional 32 g of impure product. 1H NMR (DMSO) δ ppm: 7.70 (s, 1 H), 7.37 (d, J=8.1 Hz, 1 H)1 7.09 (m, 3H), 6.5 (dd, J=1.2, 6.6 Hz, 2H), 6.38 (d, J=8.4 Hz, 1 H), 3.80 (s, 3H), 3.80 (m, 2H), 3.09-1.21 (m, 13H).
Preparation 7: (4βS,7/?,8α/?)-methyl 4β-benzyl-7-(bis(benzyloxy)phosphoryloxy)-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (4βS,7R,8αf?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β)5,6,7)8,8α,9,10-octahydrophenanthrene-2-carboxylate (5.0 g; 11.9 mmol, which may be prepared as in Preparation 6) and 5-methyltetrazole (3.6 g; 43.0 mmol) were mixed together in dichloromethane (50 mL) at ambient temperature. Dibenzylphosphoramidite (8.3 mL; 25.1 mmol) was added and the mixture was stirred until the reaction was completed (1 hour). The mixture was chilled to 00C and 30% hydrogen peroxide (10 mL) was added. The reaction was stirred until the oxidation was completed (30 minutes). The aqueous phase was separated from the organic phase. The organic phase was washed with 10% sodium meta-bisulfite (50 ml_). The organic phase was dried with anhydrous magnesium sulfate and concentrated. The crude product was purified by silica gel chromatography with 15% ethyl acetate in hexanes. The purified title compound (8.41 g; 94% yield) was obtained as a colorless oil that contained 6% ethyl acetate by weight. 1H NMR (DMSO): δ 1.31 (t, 1 H), 1.63-1.92 (m, 3H), 2.05-2.35 (m, 3H), 2.63 (d, 1 H), 2.75-3.16 (m, 4H), 3.80 (s, 3H), 5.13 (m, 4H), 6.43 (d, 1 H), 6.49 (m, 2H), 7.04-7.17 (m, 3H), 7.33-7.42 (m, 12H), 7.71 (d, 1 H).
Preparation 8: dibenzyl (2f?,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-o yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yI phosphate

The (4βS,7R,8αf?)-methyl 4β-benzyl-7-(bis(benzyloxy)phosphoryloxy)-7- (trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (7.9 g; 11.6 5 mmol, which may be prepared as in Preparation 7) and 3-amino-2-picoline (1.3 g; 12.2 mmol) were mixed together in tetrahydrofuran (80 ml_) and chilled to 0°C. The 1 M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (24 ml_; 24.4 mmol) was added while maintaining the temperature below 100C. The mixture was stirred for 30 minutes. Water (50 mL) was added to the reaction mixture. The mixture was extracted with ethyl acetate. The organic extract was washed with water. The organic phase was dried with anhydrous magnesium sulfate and concentrated. The crude product was purified by silica gel chromatography with 70% ethyl acetate in hexanes. The purified title compound (6.79 g; 68% yield) was obtained as a yellow gum that contained 6% ethyl acetate by weight. 1H NMR (DMSO): δ 1.33 (t, 1 H), 1.66-1.93 (m, 3H), 2.08-2.34 (m, 3H), 2.41 (s, 3H), 2.68 (d, 1 H), 2.76-3.19 (m, 4H), 5.14 (m, 4H), 6.47 (d, 1 H), 6.56 (m, 2H), 7.07-7.19 (m, 3H), 7.20-7.53 (m, 12H), 7.71 (d, 1 H), 7.76 (s, 1 H), 8.32 (d, 1 H), 9.93 (s, 1 H).
Example 1 : (4βS,7/?,8αR)-4β-benzyl-7-hydroxy-W-(2-methylpyridin-3-yl)-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxamide

The (4βS,7ft,8αR)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (10 g; 23.9 mmol, which may be prepared as described in Preparation 6), and 3-amino-2-picoline (2.71 g; 25.1 mmol) were dissolved in toluene (200 ml_). The 1 M lithium bis(trimethylsilyl)amide in tetrahydrofuran (74.1 mL; 74.1 mmol) was added at a rate such that the temperature was maintained below 350C. There was a mild exotherm and a solid precipitated during the addition. The mixture was held an additional 30 minutes after the addition. Water (250 mL) was added to the mixture. There was a mild exotherm and the solid dissolved. Ethyl acetate (50 mL) was added to the mixture to ensure the product did not precipitate. Stirring was stopped to allow the phases to separate. The aqueous phase was removed. The organic phase was washed with water (250 mL). Solvent (230 mL) was distilled at atmospheric pressure from the organic phase. The mixture was cooled to ambient temperature. The mixture was filtered and the solid was washed with toluene (2 times) followed by heptane (2 times). The solid was dried in a vacuum oven at 700C. The title compound of the present example (10 g) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.32 (m, 1 H), 1.82 (m, 4H), 2.10 (m, 4H), 2.41 (s, 3H), 2.68 (d, 1 H), 3.08 (m, 3H), 6.00 (s, 1H), 6.43 (d, 1 H), 6.59 (m, 2H), 7.12 (m, 3H), 7.25 (dd, 1H), 7.44 (dd, 1H), 7.71 (dd, 1 H), 7.75 (d, 1 H), 8.31 (dd, 1 H), 9.91 (s, 1 H).
Example 2: (2f?,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-i ,2,3,4,4α,9,10,1 Oα-octahydrophenanthren-2-yl dihydrogen phosphate

The dibenzyl (2R,4αS, 10αR)-4α-bθnzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl phosphate (6 g; 7.9 mmol, which may be prepared as described in Preparation 8) was dissolved in methanol (120 ml_). 5% palladium on carbon (63% water) (1.3 g; 0.4 mmol) was added to the mixture. The mixture was treated with hydrogen (50 psi) at room temperature. The reaction stalled with 12% of the monobenzylic intermediate remaining. The mixture was filtered through a pad of Celite®. Fresh catalyst (1.3 g) was added to the solution and resubmitted to the hydrogenation conditions. Once the reaction was completed, the mixture was filtered through a pad of Celite®. The solution was concentrated to about 60 ml_ by distillation and not by using a rotary evaporator. During the distillation a white solid precipitated. The mixture was cooled to ambient temperature. The mixture was filtered and the solid washed with methanol. The solid was dried in a vacuum oven at 700C. The compound of the present example (3.36 g; 75% yield) was obtained as a white solid and had an LC purity of 98 area %. 1H NMR (DMSO): δ 1.33 (t, 1 H)1 1.69-1.98 (m, 3H), 2.07-2.29 (m, 3H)1 2.42 (s, 3H), 2.61-2.80 (m, 2H)1 2.93-3.19 (m, 3H)1 3.30 (d, 1 H), 6.50 (d, 1 H), 6.64 (m, 2H), 7.08-7.20 (m, 3H), 7.29 (dd, 1 H), 7.48 (dd, 1 H), 7.75 (dd, 2H), 8.33 (dd, 1 H), 9.96 (s, 1 H).
PATENT
WO 2008093236
Example 1 : (4βS,7/?,8α/?)-4β-benzyl-7-hydroxy-N-(2-methylpyridin-3-yl)-7- (trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxamide

The (4βS,7R,8α/?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5,6J7,8,δα,9, 10- octahydrophenanthrene-2-carboxylate (10 g; 23.9 mmol, which may be prepared as described in Preparation 6), and 3-amino-2-picoline (2.71 g; 25.1 mmol) were dissolved in toluene (200 ml_). The 1 M lithium bis(trimethylsilyl)amide in tetrahydrofuran (74.1 ml_; 74.1 mmol) was added at a rate such that the temperature was maintained below 350C. There was a mild exotherm and a solid precipitated during the addition. The mixture was held an additional 30 minutes after the addition. Water (250 ml_) was added to the mixture. There was a mild exotherm and the solid dissolved. Ethyl acetate (50 ml_) was added to the mixture to ensure the product did not precipitate. Stirring was stopped to allow the phases to separate. The aqueous phase was removed. The organic phase was washed with water (250 ml_). Solvent (230 ml_) was distilled at atmospheric pressure from the organic phase. The mixture was cooled to ambient temperature. The mixture was filtered and the solid was washed with toluene (2 times) followed by heptane (2 times). The solid was dried in a vacuum oven at 700C. The title compound of the present example (10 g) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.32 (m, 1H), 1.82 (m, 4H), 2.10 (m, 4H), 2.41 (s, 3H), 2.68 (d, 1 H), 3.08 (m, 3H), 6.00 (s, 1 H), 6.43 (d, 1 H), 6.59 (m, 2H), 7.12 (m, 3H), 7.25 (dd, 1 H), 7.44 (dd, 1 H), 7.71 (dd, 1 H), 7.75 (d, 1 H), 8.31 (dd, 1 H), 9.91 (s, 1 H).
Example 2: (2f?,4αS,10α/?)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2- (trifluoromethyl)-1,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate

The dibenzyl (2R,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2- (trifluoromethyl)-1 ,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl phosphate (6 g; 7.9 mmol, which may be prepared as described in Preparation 8) was dissolved in methanol (120 ml_). 5% palladium on carbon (63% water) (1.3 g; 0.4 mmol) was added to the mixture. The mixture was treated with hydrogen (50 psi) at room temperature. The reaction stalled with 12% of the monobenzylic intermediate remaining. The mixture was filtered through a pad of Celite®. Fresh catalyst (1.3 g) was added to the solution and resubmitted to the hydrogenation conditions. Once the reaction was completed, the mixture was filtered through a pad of Celite®. The solution was concentrated to about 60 ml_ by distillation and not by using a rotary evaporator. During the distillation a white solid precipitated. The mixture was cooled to ambient temperature. The mixture was filtered and the solid washed with methanol. The solid was dried in a vacuum oven at 7O0C. The compound of the present example (3.36 g; 75% yield) was obtained as a white solid and had an LC purity of 98 area %. 1H NMR (DMSO): δ 1 .33 (t, 1 H), 1 .69- 1.98 (m, 3H), 2.07-2.29 (m, 3H), 2.42 (s, 3H), 2.61 -2.80 (m, 2H), 2.93-3.19 (m, 3H), 3.30 (d, 1 H), 6.50 (d, 1 H), 6.64 (m, 2H), 7.08-7.20 (m, 3H), 7.29 (dd, 1 H), 7.48 (dd, 1 H), 7.75 (dd, 2H), 8.33 (dd, 1 H), 9.96 (s, 1 H).
REFERENCES
https://www.pfizer.com/sites/default/files/product-pipeline/July%2028%202015%20Pipeline%20Update.pdf
////////
Cc1c(cccn1)NC(=O)c2ccc3c(c2)CC[C@H]4[C@]3(CC[C@@](C4)(C(F)(F)F)OP(=O)(O)O)Cc5ccccc5
O=P(O)(O[C@@]1(C(F)(F)F)C[C@@]2([H])CCC3=C(C=CC(C(NC4=CC=CN=C4C)=O)=C3)[C@]2(CC5=CC=CC=C5)CC1)O



.jpg)