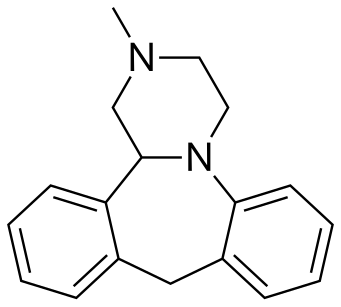
MIANSERIN
Mianserin (brand names: Depnon (IN), Lantanon (ZA), Lerivon (AR, BE, CZ, PL, RU, SK), Lumin (AU), Norval (UK), Tolvon (AU, HK†, IE†,NZ, SG†), Tolmin (DK); where † indicates discontinued products) is a psychoactive drug of the tetracyclic antidepressant (TeCA) therapeutic family. It is classified as a noradrenergic and specific serotonergic antidepressant (NaSSA) and has antidepressant,anxiolytic (anti-anxiety), hypnotic (sedating), antiemetic (nausea and vomiting-attenuating), orexigenic (appetite-stimulating), andantihistamine effects.
It is not approved for use in the US, but its analogue, mirtazapine, is. Mianserin was the first antidepressant to reach the UK market that was less dangerous than the tricyclic antidepressants in overdose.[3]

Medical uses
When used for the treatment of depression, its efficacy appears comparable to that of amitriptyline, citalopram, clomipramine,dothiepin, doxepin, fluoxetine, flupenthixol, fluvoxamine, imipramine, moclobemide, nortriptyline, paroxetine, and trazodone.[1][4]Mianserin received TGA approval in May 1996.[5]
Similarly to its analogue, mirtazapine, mianserin has been tried as an augmentation strategy in treatment-resistant depression with some success.[6] Mianserin has been tried, similarly to mirtazapine, as an adjunct in schizophrenia and has been found to reduce negative and cognitive symptoms.[7][8][9]
Mianserin has demonstrated efficacy as a monotherapy for the treatment of Parkinson's disease psychosis in an open-label clinical trial.[10]
Interactions
CYP2D6 inhibitors such as the selective serotonin reuptake inhibitors (SSRIs), quinidine, ritonavir, etc. would likely raise plasma levels of mianserin and hence could lead to mianserin toxicity. Conversely, CYP2D6 inducers would likely lead to reduced mianserin plasma concentrations and hence potentially diminish the therapeutic effects of mianserin.[1]
Withdrawal
Abrupt or rapid discontinuation of mianserin may provoke a withdrawal, the effects of which may include depression, anxiety, panic attacks,[14] decreased appetite or anorexia,insomnia, diarrhea, nausea and vomiting, and flu-like symptoms, such as allergies or pruritus, among others.
Pharmacology
Mianserin is an antagonist/inverse agonist of the H1, 5-HT1D, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT6, 5-HT7, α1-adrenergic, and α2-adrenergic receptors, and also inhibits thereuptake of norepinephrine.[16][17] As a high affinity H1 receptor inverse agonist, mianserin has strong antihistamine effects (sedation, weight gain, etc.). Contrarily, it has negligible affinity for the mACh receptors, and thus lacks anticholinergic properties. It was recently found to be a weak (Ki = 1.7 μM, EC50 = 0.53 μM) κ-opioid receptor partial agonist.[18]
In addition, mianserin also appears to be a potent antagonist of the neuronal octopamine receptor.[19] What implications this may have on mood are currently unknown, however octopamine has been implicated in the regulation of sleep, appetite and insulin production and therefore may theoretically contribute to the overall side effect profile of mianserin.[20][21]
Blockade of the H1 and α1-adrenergic receptors has sedative effects,[2] and also antagonism of the 5-HT2A and α1-adrenergic receptors inhibits activation of intracellularphospholipase C (PLC), which seems to be a common target for several different classes of antidepressants.[22] By antagonizing the somatodendritic and presynaptic α2-adrenergic receptors which function predominantly as inhibitory autoreceptors and heteroreceptors, mianserin disinhibits the release of norepinephrine, dopamine, serotonin, andacetylcholine in various areas of the brain and body.
Enantioselectivity

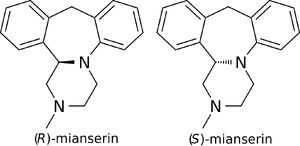
(S)-(+)-Mianserin is approximately 200–300 times more active than its enantiomer (R)-(−)-mianserin.

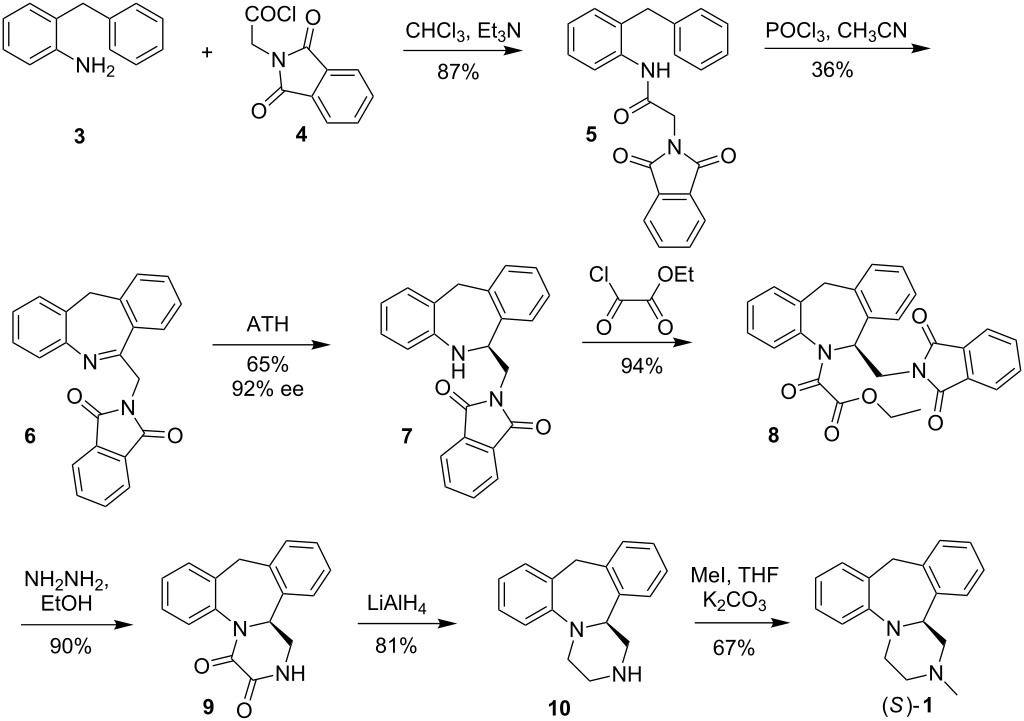
(14bS)-(+)-1,2,3,4,10,14b-Hexahydro-2-methyldibenzo[c,f]pyrazino[1,2-a]azepine (1)
(S)-(+)-1 in the form of solidifying oil; during purification step a small degree of product decomposition was observed; []D 23 = +469.2 (c 1, CHCl3); []D 23 = +436.5 (c 1, EtOH) {[9] []D 23 = +450 (c 0.26, EtOH)}; []D 23 = +428.0 (c 0.5, MeOH) {[5] []D 25 = +469.0 (c 1, MeOH)}.
Enantiomeric purity was determined by HPLC analysis (Chiracel OD-H, hexane:2- propanol = 80:20, 1ml/min, S isomer 5.6min).
IR (CCl4): 3064, 3022, 2939, 2794, 1492, 1446, 1251, 1132 cm–1 ;
1H NMR (500 MHz, CDCl3): δ 2.37-2.42 (m, 4 H), 2.46 (t, J = 10.5 Hz, 1 H), 2.92 (dt, J1 = 2.0 Hz, J2 = 11.0 Hz, 1 H), 3.02 (dd, J1 = 1.5 Hz, J2 = 11.0 Hz, 1 H), 3.25-3.28 (m, 1 H), 3.30 (d, J = 13.0 Hz, 1 H, methylene bridge), 3.42 (td, J1 = 3.0 Hz, J2 = 11.0 Hz, 1 H), 4.14 (dd, J1 = 2.0 Hz, J2 = 10.0 Hz, 1 H,methine), 4.81 (d, J = 13.0 Hz, 1 H, methylene bridge), 6.87 (td, J1 = 1.0 Hz, J2 = 7.5 Hz, 1 H, Ar), 7.00-7.02 (m, 2 H, Ar), 7.05-7.13 (m, 4 H, Ar), 7.16 (td, J1 = 1.5 Hz, J2 = 7.5 Hz, 1 H, Ar);
13C NMR (125 MHz, CDCl3): δ 38.8, 45.6, 51.0, 55.4, 64.6, 66.2, 119.0, 122.3, 126.5, 126.6, 127.0, 127.3, 128.1, 129.5, 137.1, 139.3, 139.8, 148.4.
HRMS (ESI): m/z calcd for C18H21N2 [M+H]+ : 265.1705; found: 265.1712.
(±)-1,2,3,4,10,14b-Hexahydro-2-methyldibenzo[c,f]pyrazino[1,2-a]azepine (1)
The racemate was prepared in the same manner as pure enantiomer; mp = 109.5- 110.5 °C ([10] mp = 111–113 °C). The HPLC analysis (Chiracel OD-H, hexane/2- propanol = 80:20, 1mL/min, R isomer 5.0 min and S isomer 5.6 min)
SYN 1
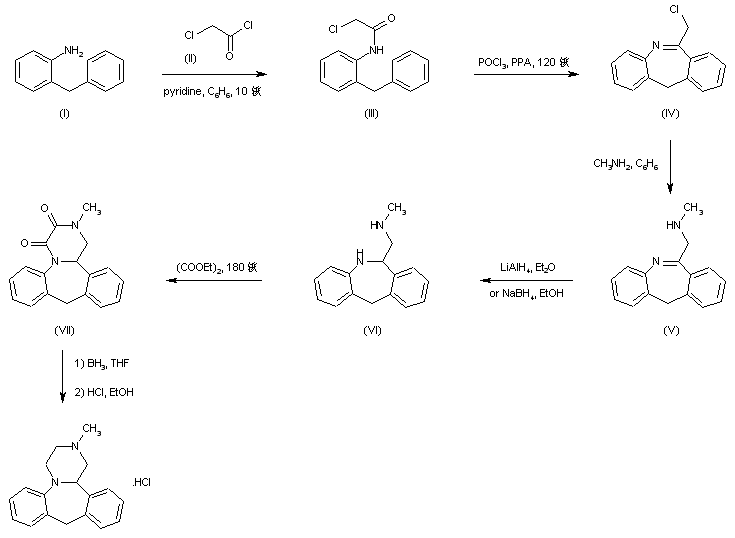
The title compound has been synthesized by several procedures. Acylation of 2-benzylaniline (I) by chloroacetyl chloride (II) gave chloroacetamide (III). Subsequent cyclization of amide (III) under Vilsmeier conditions furnished the dibenzoazepine (IV). Nucleophilic substitution of the chlorine atom of (IV) by methylamine led to amine (V). The imine function of (V) was reduced with either LiAlH4 or NaBH4 to the diamine (VI), which was further converted into the fused diketopiperazine (VII) upon heating with diethyl oxalate. The amide groups of (VII) were then reduced by means of borane in THF, yielding the target tetracyclic diamine, which was finally isolated as the corresponding hydrochloride salt......US 3534041
SYN 2
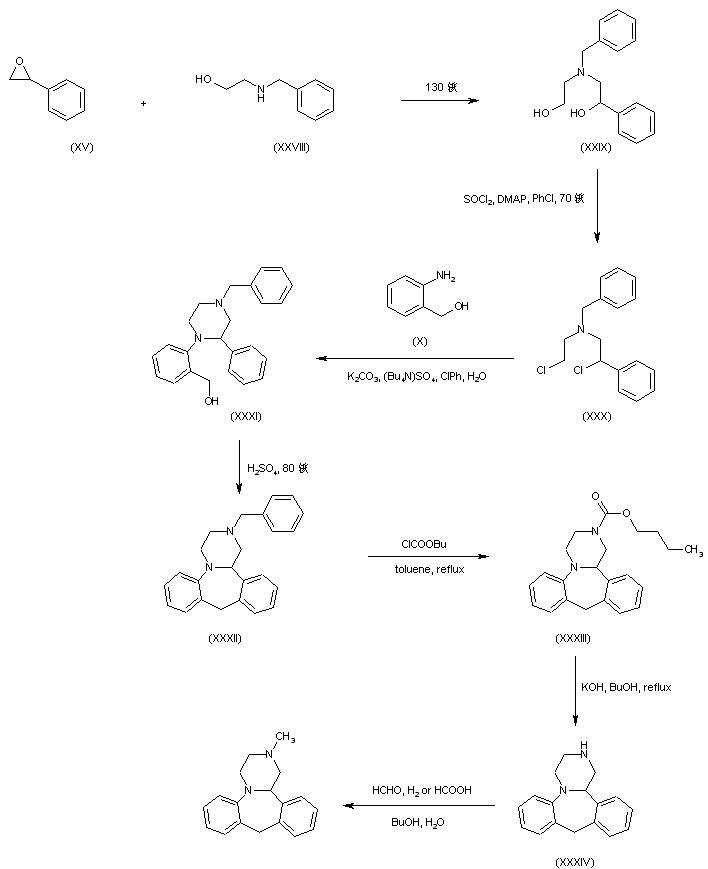
In a further procedure, styrene oxide (XV) was condensed with 2-(benzylamino)ethanol (XXVIII) to give amino diol (XXIX). After chlorination of (XXIX) using SOCl2 and DMAP, dichloro derivative (XXX) was condensed with 2-aminobenzyl alcohol (X) yielding piperazine (XXXI). Cyclization of (XXXI) in hot sulfuric acid afforded the tetracyclic compound (XXXII). The N-benzyl group of (XXXII) was then removed by treatment with butyl chloroformate producing carbamate (XXXIII), which was further hydrolyzed and decarboxylated to (XXXIV) under basic conditions. Finally, methylation of the secondary amine (XXXIV) was performed by reductive alkylation with formaldehyde either in the presence of formic acid under Leuckart-Wallach conditions or by catalytic hydrogenation
DE 4305659; EP 0612745
SYN 3
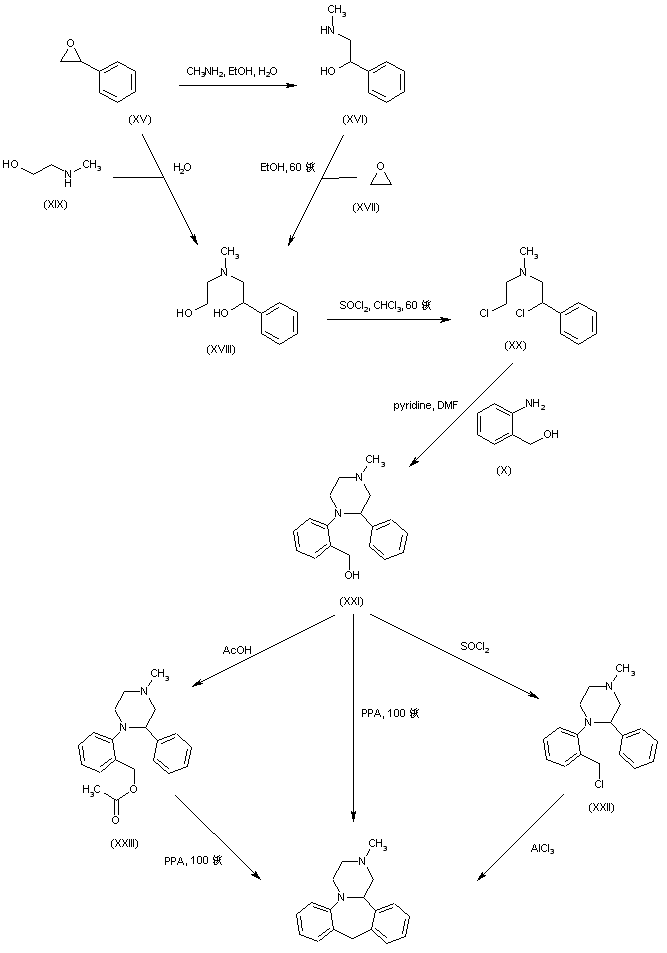
In a different method, reaction of styrene oxide (XV) with methylamine provided amino alcohol (XVI), which was further condensed with ethylene oxide (XVII) to afford amino diol (XVIII). Alternatively, diol (XVIII) was prepared by a more direct procedure by condensation of epoxide (XV) with 2-(methylamino)ethanol (XIX). Chlorination of (XVIII) employing SOCl2 yielded the dichloro derivative (XX), which was subsequently condensed with 2-aminobenzyl alcohol (X) leading to piperazine (XXI). Cyclization of (XXI) to the title compound was accomplished by treatment with hot polyphosphoric acid. Optionally, alcohol (XXI) was converted to chloride (XXII), which was then cyclized in the presence of AlCl3. In a related method, alcohol (XXI) was esterified with AcOH, and the resultant acetate ester (XXIII) was then cyclized in the presence of polyphosphoric acid......US 4217452
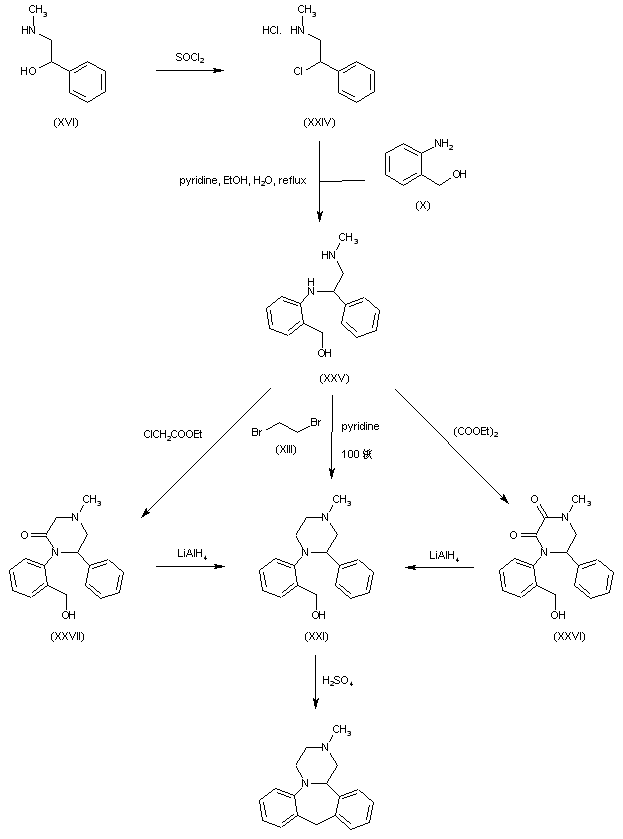
The key intermediate (XXI) was also prepared by several related procedures. Chlorination of aminoalcohol (XVI) gave chloro amine (XXIV), which was condensed with 2-aminobenzyl alcohol (X) to afford diamine (XXV). Then, alkylation of diamine (XXV) with dibromoethane (XIII) in hot pyridine gave rise to the target piperazine (XXI). Alternatively, diamine (XXV) was condensed with ethyl chloroacetate or with diethyl oxalate to produce the mono- or dioxopiperazines (XXVII) and (XXVI), respectively, which were then reduced to (XXI) by means of LiAlH4. Cyclization of alcohol (XXI) to the title compound was achieved by treatment with concentrated sulfuric acid
SYN5
FR 2647114
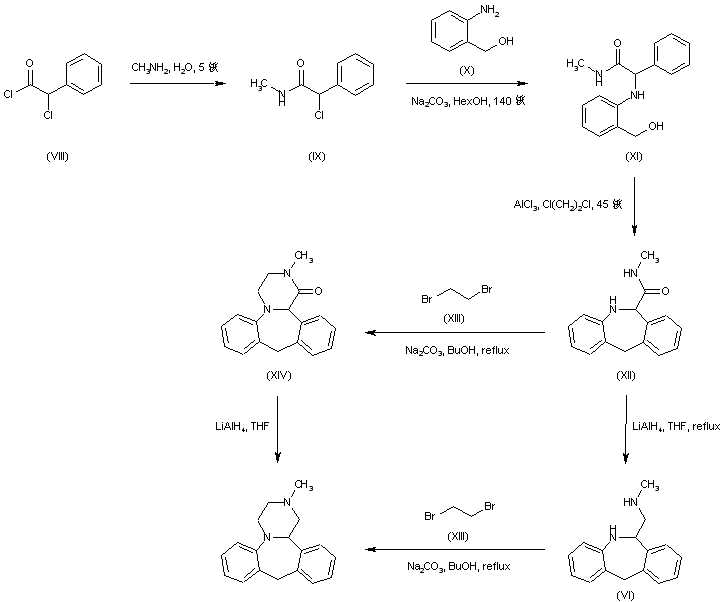
Treatment of alpha-chlorophenylacetyl chloride (VIII) with methylamine provided the corresponding chloro amide (IX), which was subsequently condensed with 2-aminobenzyl alcohol (X) to afford amino alcohol (XI). Cyclization of (XI) in the presence of AlCl3 led to the dibenzoazepine (XII). This was converted to the tetracyclic compound (XIV) by reaction with dibromoethane (XIII) in the presence of Na2CO3. Reduction of the amide carbonyl group of (XIV) by means of LiAlH4 furnished the title compound. In a related strategy, amide (XII) was initially reduced to diamine (VI) by using LiAlH4. Subsequent condensation of (VI) with dibromoethane (XIII) led to the target tetracyclic derivative
OTHER..........


References
- Truven Health Analytics, Inc. DRUGDEX® System (Internet) [cited 2013 Sep 29]. Greenwood Village, CO: Thomsen Healthcare; 2013.
- Merck Sharp & Dohme (Australia) Pty Limited. "Tolvon Product Information"(PDF). GuildLink Pty Ltd.
- Walker, R; Whittlesea, C, ed. (2007) [1994]. Clinical Pharmacy and Therapeutics (4th ed.). Edinburgh: Churchill Livingstone Elsevier. ISBN 978-0-7020-4293-5.
- Wakeling A (April 1983). "Efficacy and side effects of mianserin, a tetracyclic antidepressant". Postgrad Med J 59 (690): 229–31. doi:10.1136/pgmj.59.690.229.PMC 2417496. PMID 6346303.
- AlphaPharm. "Lumin Mianserin hydrochloride product information" (PDF). GuildLink Pty Ltd.
- Ferreri M, Lavergne F, Berlin I, Payan C, Puech AJ (January 2001). "Benefits from mianserin augmentation of fluoxetine in patients with major depression non-responders to fluoxetine alone". Acta Psychiatr Scand 103 (1): 66–72. doi:10.1111/j.1600-0447.2001.00148.x. PMID 11202131.
- Poyurovsky, M; Koren, D; Gonopolsky, I; Schneidman, M; Fuchs, C; Weizman, A; Weizman, R (March 2003). "Effect of the 5-HT2 antagonist mianserin on cognitive dysfunction in chronic schizophrenia patients: an add-on, double-blind placebo-controlled study". European Neuropsychopharmacology 13 (2): 123–128. doi:10.1016/S0924-977X(02)00155-4. PMID 12650957.
- Shiloh, R; Zemishlany, Z; Aizenberg, D; Valevski, A; Bodinger, L; Munitz, H; Weizman, A (March 2002). "Mianserin or placebo as adjuncts to typical antipsychotics in resistant schizophrenia". International Clinical Psychopharmacology 17 (2): 59–64.doi:10.1097/00004850-200203000-00003. PMID 11890187.
- Mizuki, Y; Kajimura, N; Imai, T; Suetsugi, M; Kai, S; Kaneyuki, H; Yamada, M (April 1990). "Effects of mianserin on negative symptoms in schizophrenia". International Clinical Psychopharmacology 5 (2): 83–95. doi:10.1097/00004850-199004000-00002.PMID 1696292.
- Ikeguchi, K; Kuroda, A (1995). "Mianserin treatment of patients with psychosis induced by antiparkinsonian drugs". European Archives of Psychiatry and Clinical Neuroscience 244(6): 320–324. doi:10.1007/BF02190411. PMID 7772616.
- "Australian Medicines Handbook". Australian Medicines Handbook Pty Ltd. 2013.
- British National Formulary (BNF) (65th ed.). Pharmaceutical Press. p. 1120.ISBN 978-0857110848.
- Mianserin Hydrochloride. Martindale: The Complete Drug Reference (The Royal Pharmaceutical Society of Great Britain). 5 December 2011. Retrieved 3 November 2013.
- Kuniyoshi M, Arikawa K, Miura C, Inanaga K (June 1989). "Panic anxiety after abrupt discontinuation of mianserin". Jpn. J. Psychiatry Neurol. 43 (2): 155–9. doi:10.1111/j.1440-1819.1989.tb02564.x. PMID 2796025.
- Taylor D, Paton C, Kapur S, Taylor D. The Maudsley prescribing guidelines in psychiatry. 11th ed. Chichester, West Sussex: John Wiley & Sons; 2012.
- Leonard B, Richelson H (2000). "Synaptic Effects of Antidepressants: Relationship to Their Therapeutic and Adverse Effects". In Buckley JL, Waddington PF. Schizophrenia and Mood Disorders: The New Drug Therapies in Clinical Practice. Oxford: Butterworth-Heinemann. pp. 67–84. ISBN 978-0-7506-4096-1.
- Müller G (8 May 2006). "Target Family-directed Masterkeys in Chemogenomics". In Kubinyi H, Müller G, Mannhold R, Folkers G. Chemogenomics in Drug Discovery: A Medicinal Chemistry Perspective. John Wiley & Sons. p. 25. ISBN 978-3-527-60402-9. Retrieved 13 May 2012.
- Olianas MC, Dedoni S, Onali P (November 2012). "The atypical antidepressant mianserin exhibits agonist activity at κ-opioid receptors". Br. J. Pharmacol. 167 (6): 1329–41.doi:10.1111/j.1476-5381.2012.02078.x. PMID 22708686.
- Roeder T (November 1990). "High-affinity antagonists of the locust neuronal octopamine receptor". Eur. J. Pharmacol. 191 (2): 221–4. doi:10.1016/0014-2999(90)94151-M.PMID 2086239.
- Crocker A, Sehgal A (September 2008). "Octopamine regulates sleep in drosophila through protein kinase A-dependent mechanisms". J. Neurosci. 28 (38): 9377–85.doi:10.1523/JNEUROSCI.3072-08a.2008. PMC 2742176. PMID 18799671.
- Bour S, Visentin V, Prévot D, Carpéné C (September 2003). "Moderate weight-lowering effect of octopamine treatment in obese Zucker rats". J. Physiol. Biochem. 59 (3): 175–82.doi:10.1007/BF03179913. PMID 15000448.
- Dwivedi Y, Agrawal AK, Rizavi HS, Pandey GN (December 2002). "Antidepressants reduce phosphoinositide-specific phospholipase C (PI-PLC) activity and the mRNA and protein expression of selective PLC beta 1 isozyme in rat brain". Neuropharmacology 43(8): 1269–79. doi:10.1016/S0028-3908(02)00253-8. PMID 12527476.
- Roth, BL; Driscol, J (12 January 2011). "PDSP Ki Database". Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 13 October 2013.
Further reading
- Peet M, Behagel H (1978). "Mianserin: a decade of scientific development". Br. J. Clin. Pharmacol. 5 Suppl 1: 5S–9S. PMC 1429213. PMID 623702.
External links
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
| (±)-2-methyl-1,2,3,4,10,14b-hexahydrodibenzo[c,f]pyrazino[1,2-a]azepine | |
| Clinical data | |
| Trade names | Bolvidon (discontinued), Tolvon |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Legal status | |
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 20–30%[1] |
| Protein binding | 95%[1] |
| Metabolism | Hepatic (mediated byCYP2D6; most metabolism occurs via aromatic hydroxylation, N-oxidation and N-demethylation)[1] |
| Biological half-life | 21–61 hours[2] |
| Excretion | Renal (4–7%) Faecal (14–28%)[1] |
| Identifiers | |
| CAS Number | 24219-97-4 |
| ATC code | N06AX03 |
| PubChem | CID 4184 |
| IUPHAR/BPS | 135 |
| DrugBank | DB06148 |
| ChemSpider | 4040 |
| UNII | 250PJI13LM |
| KEGG | D08216 |
| ChEBI | CHEBI:51137 |
| ChEMBL | CHEMBL6437 |
| Chemical data | |
| Formula | C18H20N2 |
| Molar mass | 264.365 |
///////////MIANSERIN
c42c(N3C(c1ccccc1C2)CN(C)CC3)cccc4