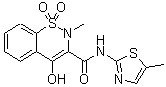
Meloxicam
351.40, C14H13N3O4S2, MP 255 °C
(8E)-8-[hydroxy-[(5-methyl-1,3-thiazol-2-yl)amino]methylidene]-9-methyl-10,10-dioxo-10$l^{6}-thia-9-azabicyclo[4.4.0]deca-1,3,5-trien-7-one;
4-Hydroxy-2-methyl-N-(5-methyl-2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxide;
CAS 133687-22-6; Mobec;Mobic (TN);
2H-1,2-Benzothiazine-3-carboxamide, 4-hydroxy-2-methyl-N-(5-methylthiazolyl)-, 1,1-dioxide;
The IUPAC name of Meloxicam is (3E)-3-[hydroxy-[(5-methyl-1,3-thiazol-2-yl)amino]methylidene]-2-methyl-1,1-dioxo-1λ6,2-benzothiazin-4-one. With the CAS registry number 71125-38-7, it is also named as 2H-1,2-Benzothiazine-3-carboxamide, 4-hydroxy-2-methyl-N-(5-methylthiazolyl)-, 1,1-dioxide.
Uses of Meloxicam: this chemical is a nonsteroidal anti-inflammatory drug with analgesic and fever reducer effects. And it inhibits cyclooxygenase that can be used as an anti-inflammatory. Additionally, it can be used for the treatment of rheumatoid arthritis and osteoarthritis.
In Europe, where the product has been available since the early 1990s, it is also prescribed and licensed for other anti-inflammatory benefits including relief from both acute and chronic pain in dogs and cats. For many years, both injectable and oral (liquid and tablet) formulations of meloxicam have been licensed for use in dogs, and injectable ones for use in cats. In June 2007, a new oral version of Metacam was licensed in Europe for the long-term relief of pain in cats. As of June 2008, Meloxicam is registered for long term use in cats in Australia, New Zealand, and throughout Europe. 'Metacam oral suspension 1.5 is not approved or recommended (according to the manufacture insert) for use in cats in the U.S.

1H NMR DMSOD6


13C NMR DMSOD6
Meloxicam is a nonsteroidal anti-inflammatory drug (NSAID) with analgesic and fever reducer effects. It is a derivative of oxicam, closely related to piroxicam, and falls in the enolic acid group of NSAIDs.[2] It was developed by Boehringer-Ingelheim. Meloxicam starts to relieve pain about 30–60 minutes after administration.[3]
Mechanism of action
Main article: Non-steroidal anti-inflammatory drug
Meloxicam blocks cyclooxygenase (COX), the enzyme responsible for converting arachidonic acid into prostaglandin H2—the first step in the synthesis of prostaglandins, which are mediators of inflammation. Meloxicam has been shown, especially at its low therapeutic doses, selectively to inhibit COX-2 over COX-1.[1]
Meloxicam concentrations in synovial fluid range from 40% to 50% of those in plasma. The free fraction in synovial fluid is 2.5 times higher than in plasma, due to the lower albumin content in synovial fluid as compared to plasma. The significance of this penetration is unknown,[2] but it may account for the fact that it performs exceptionally well in treatment of arthritis in animal models.[4]
Side effects
Meloxicam use can result in gastrointestinal toxicity and bleeding, headaches, rash, and very dark or black stool (a sign of intestinal bleeding). Like other NSAIDs, its use is associated with an increased risk of cardiovascular events such as heart attack and stroke.[5]It has fewer gastrointestinal side effects than diclofenac,[6] piroxicam,[7] naproxen,[8] and perhaps all other NSAIDs which are not COX-2 selective.[6] Although meloxicam does inhibit thromboxane A, it does not appear to do so at levels that would interfere withplatelet function.
A pooled analysis of randomized, controlled studies of meloxicam therapy of up to 60 days duration found that meloxicam was associated with a statistically significantly lower number of thromboembolic complications than the NSAID diclofenac (0.2% versus 0.8% respectively) but a similar incidence of thromboembolic events to naproxen and piroxicam.[9]
Potential serious cardiovascular side effects
Persons with hypertension, high cholesterol, or diabetes are at risk for cardiovascular side effects. Persons with family history of heart disease, heart attack or stroke must tell their treating physician as the potential for serious cardiovascular side effects is significant.[10][11]
Veterinary use
Meloxicam is also used in the veterinary field, most commonly in dogs and cats, but also sees off-label use in other animals such as cattle and exotics.[12][13] The U.S. Food and Drug Administration sent a Notice of Violation to the manufacturer for its promotional materials which included promotion of the drug for off-label use.[14] In the U.S. the drug is indicated for management of pain and inflammation associated with osteoarthritis in dogs only. In Europe, where the product has been available since the early 1990s,[citation needed] it is also prescribed and licensed for other anti-inflammatory benefits including relief from both acute and chronic pain in dogs. Side effects in animals are similar to those found in humans; the principal side effect is gastrointestinal irritation (vomiting, diarrhea and ulceration). Rarer but important side effects include liver and kidney toxicity.
Since 2003, the oral (liquid) formulations of meloxicam have been licensed in the U.S for use in dogs only,[15] with the January 2005 product insert specifically warning in bold-face type: "Do not use in cats."[16] An injectable formulation for use in dogs was approved by the FDA in November 2003,[17] with a formulation for cats, for surgical use only, approved in October 2004.[18]
In the U.S., per the manufacturer's clinical instructions as of July 2010, injectable meloxicam is indicated in operative use with felines as a single, one-time dose only, with specific and repeated warnings not to administer a second dose.[19] In June 2007, a new oral version of meloxicam was licensed in Europe for the long-term relief of pain in cats. As of June 2008, meloxicam is registered for long term use in cats in Australia, New Zealand, and throughout Europe. A peer-reviewed journal article cites feline overdose of NSAIDs, including meloxicam, as being a cause of severe kidney damage in cats.[20]
Meloxicam has been investigated as an alternative to Diclofenac by the RSPB to prevent deaths of vultures.
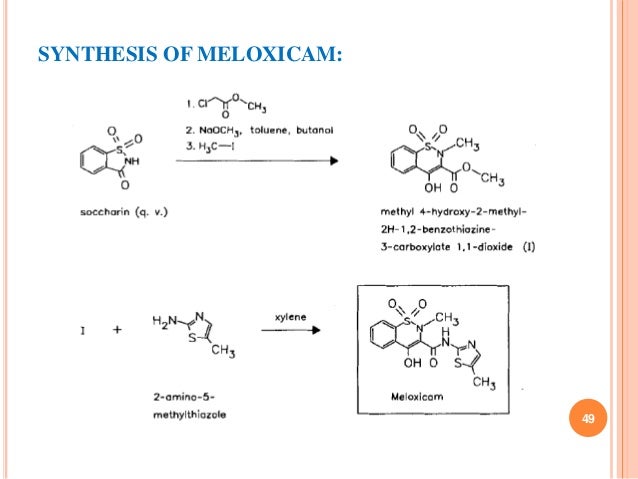
Preparation of Meloxicam: this chemical can be prepared by Methyl 4-hydroxy-2-methyl-(2H)-1,2-benzothiazine-3-carboxylate-1,1-dioxide and 2-Amino-5-methylthiazole. The yield is 74 %.
.jpg)

References
- Noble, S; Balfour, JA (March 1996). "Meloxicam.". Drugs 51 (3): 424–30; discussion 431–32. doi:10.2165/00003495-199651030-00007. PMID 8882380.
- "Meloxicam official FDA information, side effects, and uses". Drugs.com. March 2010. Retrieved 17 March 2010.
- Auvinet, B; Ziller, R; Appelboom, T; Velicitat, P (November–December 1995). "Comparison of the onset and intensity of action of intramuscular meloxicam and oral meloxicam in patients with acute sciatica.". Clinical Therapeutics 17 (6): 1078–98.doi:10.1016/0149-2918(95)80086-7. PMID 8750399.
- Engelhardt, G; Homma, D; Schlegel, K; Utzmann, R; Schnitzler, C (Oct 1995). "Anti-inflammatory, analgesic, antipyretic and related properties of meloxicam, a new non-steroidal anti-inflammatory agent with favourable gastrointestinal tolerance". Inflammation Research 44 (10): 423–433. doi:10.1007/BF01757699. PMID 8564518.
- Stamm O, Latscha U, Janecek P, et al. (January 1976). "Development of a special electrode for continuous subcutaneous pH measurement in the infant scalp". Am. J. Obstet. Gynecol. 124 (2): 193–5. PMID 2012.
- Hawkey, C; Kahan, A; Steinbrü, K; Alegre, C; Baumelou, E; Bégaud, B; Dequeker, J; Isomäki, H; et al. (Sep 1998). "Gastrointestinal tolerability of meloxicam compared to diclofenac in osteoarthritis patients". Rheumatology 37 (9): 937–945(9).doi:10.1093/rheumatology/37.9.937.
- Dequeker, J; Hawkey, C; Kahan, A; Steinbruck, K; Alegre, C; Baumelou, E; Begaud, B; Isomaki, H; et al. (1998). "Improvement in gastrointestinal tolerability of the selective cyclooxygenase (COX)-2 inhibitor, meloxicam, compared with piroxicam: results of the Safety and Efficacy Large-scale Evaluation of COX- inhibiting Therapies (SELECT) trial in osteoarthritis". The British Journal of Rheumatology 37 (9): 946–51.doi:10.1093/rheumatology/37.9.946. PMID 9783758.
- Wojtulewski, JA; Schattenkirchner, M; Barceló, P; Le Loët, X; Bevis, PJR; Bluhmki, E; Distel, M. "A Six-Month Double-Blind Trial to Compare the Efficacy and Safety of Meloxicam 7.5 mg Daily and Naproxen 750 mg Daily in Patients with Rheumatoid Arthritis".Rheumatology. 35, Supplement 1: 22–8. doi:10.1093/rheumatology/35.suppl_1.22.
- Singh, G; Lanes, S; Steinbrü, G; Triadafilopoulos (2004). "Gastrointestinal tolerability of meloxicam compared to diclofenac in osteoarthritis patients". Am J Med 117 (9): 100–6.doi:10.1016/j.amjmed.2004.03.012. PMID 15234645.
- "Medline Plus". Nlm.nih.gov. Retrieved 15 November 2014.
- "Drugs.com". Drugs.com. Retrieved 15 November 2014.
- Off-label use discussed in: Arnold Plotnick MS, DVM, ACVIM, ABVP, Pain Management using Metacam, and Stein, Robert, Perioperative Pain Management Part IV, Looking Beyond Butorphanol, Sep 2006, Veterinary Anesthesia & Analgesia Support Group.
- For off-label use example in rabbits, see Krempels, Dana, Hind Limb Paresis and Paralysis in Rabbits, University of Miami Biology Department.
- US FDA Notice of Violation for off-label use promotion, April 2005.
- "NADA 141-213: New Animal Drug Application Approval (for Metacam (meloxicam) 0.5 mg/mL and 1.5 mg/mL Oral Suspension)" (PDF). US Food and Drug Administration. April 15, 2003. Retrieved 24 July 2010.
- Metacam Client Information Sheet, product description: "Non-steroidal anti-inflammatory drug for oral use in dogs only", and in the "What Is Metacam" section in bold-face type: "Do not use in cats.", January 2005.
- "Metacam 5 mg/mL Solution for Injection" (PDF). Fda.gov. Retrieved 15 November2014.
- "Metacam 5 mg/mL Solution for Injection, Supplemental Approval" (PDF). Fda.gov. October 28, 2004. Retrieved 15 November 2014.
- See the manufacturer's FAQ on its website, and its clinical dosing instructions for cats.
- Merola, Valentina, DVM, DABT, and Dunayer Eric, MS, VMD, DABT, The 10 most common toxicoses in cats, Toxicology Brief, Veterinary Medicine, pp. 340–342, June, 2006.
- Kimble, B.; Black, L. A.; Li, K. M.; Valtchev, P.; Gilchrist, S.; Gillett, A.; Higgins, D. P.; Krockenberger, M. B.; Govendir, M. (2013). "Pharmacokinetics of meloxicam in koalas ( ) after intravenous, subcutaneous and oral administration". Journal of Veterinary Pharmacology and Therapeutics 36 (5): 486–493. doi:10.1111/jvp.12038.PMID 23406022.
External links
- Manufacturer's Official Product Website for the Veterinary formulations
- Manufacturer's United States Division website for the Veterinary formulations
- FDA Metacam
- Meloxicam Side Effects
 | |
| Systematic (IUPAC) name | |
|---|---|
| 4-hydroxy-2-methyl-N-(5-methyl-2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide-1,1-dioxide. | |
| Clinical data | |
| Trade names | Mobic |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a601242 |
| Pregnancy category | |
| Legal status | |
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 89%[1] |
| Protein binding | 99.4%[1] |
| Metabolism | Hepatic (CYP2C9 and 3A4-mediated)[1] |
| Biological half-life | 20 hours[1] |
| Excretion | Urine and faeces equally[1] |
| Identifiers | |
| CAS Number | 71125-38-7 |
| ATC code | M01AC06 |
| PubChem | CID 5281106 |
| IUPHAR/BPS | 7220 |
| DrugBank | DB00814 |
| ChemSpider | 10442740 |
| UNII | VG2QF83CGL |
| KEGG | D00969 |
| ChEBI | CHEBI:6741 |
| ChEMBL | CHEMBL599 |
| PDB ligand ID | MXM (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C14H13N3O4S2 |
| Molar mass | 351.403 g/mol |
/////
Cc1cnc(s1)NC(=O)C\3=C(/O)c2ccccc2S(=O)(=O)N/3C