 .
.Picture credit....Bethany Halford

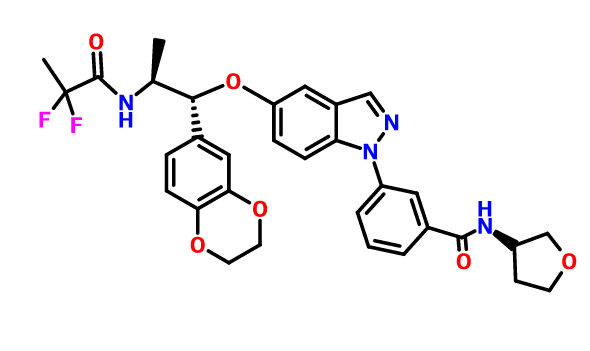
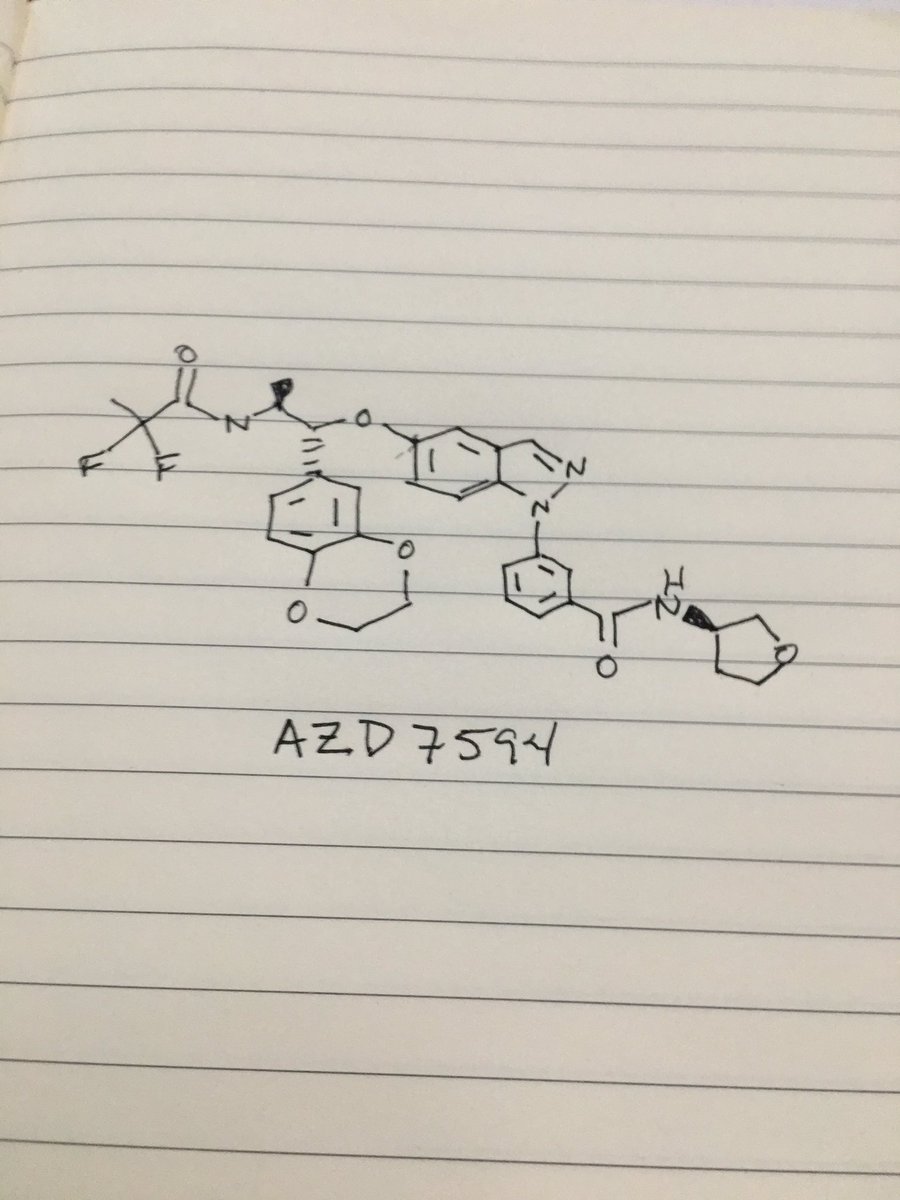
AZD 7594
AZ13189620; AZD-7594
Bayer Pharma Aktiengesellschaft, Astrazeneca Ab
| Molecular Formula: | C32H32F2N4O6 |
|---|---|
| Molecular Weight: | 606.616486 g/mol |
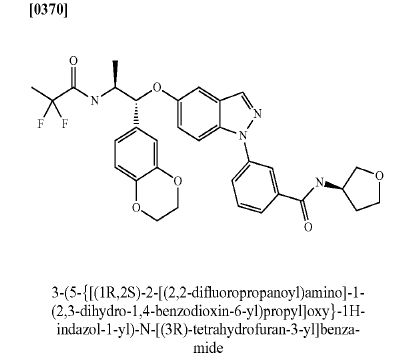
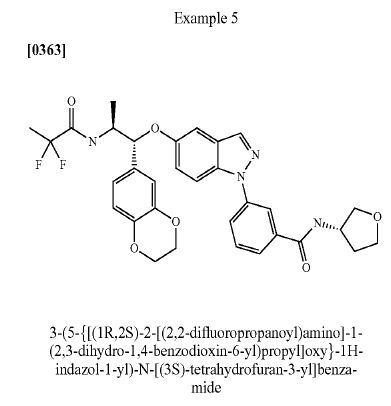
Benzamide, 3-[5-[(1R,2S)-2-[(2,2-difluoro-1-oxopropyl)amino]-1-(2,3-dihydro-1,4-benzodioxin-6-yl)propoxy]-1H-indazol-1-yl]-N-[(3R)-tetrahydro-3-furanyl]-
- Cas 1196509-60-0
It is also in phase I clinical trials for the treatment of chronic obstructive pulmonary disorder (COPD).
https://clinicaltrials.gov/ct2/show/NCT02479412
| Company | AstraZeneca plc |
| Description | Inhaled selective glucocorticoid receptor (GCCR) modulator |
| Molecular Target | Glucocorticoid receptor (GCCR) |
- Phase II Asthma
- Phase I Chronic obstructive pulmonary disease
- 01 Feb 2016 AstraZeneca completes a phase II trial in Asthma in Bulgaria and Germany (Inhalation) (NCT02479412)
- 09 Jan 2016 AstraZeneca plans to initiate a phase I trial in Healthy volunteers in USA (IV and PO) (NCT02648438)
- 01 Jan 2016 Phase-I clinical trials in Chronic obstructive pulmonary disease (In volunteers) in USA (PO, IV, Inhalation) (NCT02648438)
http://www.google.com/patents/WO2009142569A1
PATENT
US20100804345
UNWANTED ISOMER

WANTED COMPD


PATENT
WO 2009142571Example 6
WANTED ISOMER

UNWANTED

APCI-MS: m/z 607 [MH+]
1H NMR (400 MHz, DMSO-J6) δ 8.71 (IH, d), 8.65 (IH, d), 8.24 (IH, s), 8.18 (IH, s),
7.90 - 7.84 (2H, m), 7.77 (IH, d), 7.65 (IH, t), 7.21 (IH, dd), 7.13 (IH, d), 6.89 - 6.78 (3H,s m), 5.17 (IH, d), 4.48 (IH, m), 4.24 - 4.11 (5H, m), 3.90 - 3.81 (2H, m), 3.72 (IH, td), 3.61
(IH, dd), 2.16 (IH, m), 1.94 (IH, m), 1.55 (3H, t), 1.29 (3H, d).
LC (Method A) rt = 12.02 min
LC (Method B) rt = 11.12 min
Chiral SFC (method B) rt = 5.10 min o M.p. = 175 0C
PATENT
WO 2011061527http://www.google.com/patents/WO2011061527A1?cl=en
Intermediate 12
( 1 R,2S)-2-amino- 1 -(2,3 -dihydrobenzo b [ 1 ,41dioxin-6-yl)propan- 1 -ol hydrochloride. (12)

APCI-MS: m/z 210 [MH+-HC1]
1H-NMR (300 MHz, DMSO-^): δ 8.01 (brs, 3H), 6.87-6.76 (m, 3H), 5.93 (brd, 1H), 4.79 (brt, 1H), 4.22 (s, 4H), 3.32 (brm, 1H), 0.94 (d, 3H).
tert-butyl (1R,2S)- 1 -(2,3-dihvdrobenzorbl Γ 1 ,41dioxin-6-yl)- 1 -hvdroxypropan-2-ylcarbamate.

(S)-tert-butyl 1 -(2,3-dihydrobenzo[b] [ 1 ,4]dioxin-6-yl)- 1 -oxopropan-2-ylcarbamate (I2b) (3.76 g, 12.23 mmol), aluminium isopropoxide (0.5 g, 2.45 mmol) and 2-propanol (12 mL, 157.75 mmol) in toluene (22 mL) were stirred at 50°C under argon for 16 hours. The reaction mixture was poured into 1M HC1 (150 mL) and the mixture was extracted with ethyl acetate (250 mL). The organic phase was washed with water (2x50 mL) and brine (100 mL), dried over Na2SC"4, filtered and concentrated. The crude product was purified by flash- chromatography on silica using ethyl acetate/hexane (1/2) as eluent. Fractions containing product were combined. Solvent was removed by evaporation to give the desired product as a colourless solid. Yield 3.19 g (84%) APCI-MS: m/z 236, 210, 192 [MH -tBu-18, MH -BOC, MH -BOC- 18]
1H NMR (300 MHz, DMSO-^): δ 6.80-6.70 (m, 3H), 6.51 (d, IH), 5.17 (d, IH), 4.36 (t, IH),
4.19 (s, 4H), 3.49 (m, IH), 1.31 (s, 9H), 0.93 (d, 3H).
(S)-tert-butyl 1 -(2,3-dihydrobenzo[bl [ 1 ,41dioxin-6-yD- 1 -oxopropan-2-ylcarbamate. (I2b)

(95%)
APCI-MS: m/z 208 [MH+ - BOC]
1H NMR (300 MHz, DMSO-^): δ 7.50 (dd, IH), 7.46 (d, IH), 7.24 (d, IH), 6.97 (d, IH), 4.97 (m, IH), 4.30 (m, 4H), 1.36 (s, 9H), 1.19 (d, 3H).
Intermediate 13
(lR,2S)-2-amino-l-(4H-benzo[dl[l,31dioxin-7- l)propan-l-ol hydrochloride (13)

APCI-MS: m/z 210 [MH+ -HC1]
1H NMR (300 MHz, DMSO-^) δ 8.07 (3H, s), 7.05 (IH, d), 6.92 (IH, dd), 6.85 (IH, d), 6.03 (IH, d), 5.25 (2H, s), 4.87 (3H, m), 3.42 - 3.29 (IH, m), 0.94 (3H, d).
(4S.5R -5-(4H-benzordiri.31dioxin-7-vn- -methyloxazolidin-2-one (I3a

The relative cis conformation of the product was confirmed by comparing the observed 1H- NMR with the literature values reported for similar cyclised norephedrine (Org. Lett. 2005 (07), 13, 2755-2758 and Terahedron Assym. 1993, (4), 12, 2513-2516). In a 2D NOESY experiment a strong NOE cross-peak was observed for the doublet at 5.64 with the multiplet at 4.19 ppm. This also confirmed the relative czs-conformation.
APCI-MS: m/z 236 [MH+]
1H NMR (400 MHz, CDC13) δ 6.99 (d, J= 8.0 Hz, IH), 6.88 (dd, J= 8.0, 1.4 Hz, IH), 6.83 (s, IH), 5.81 (brs,lH), 5.64 (d, J= 8.0 Hz, IH), 5.26 (s, 2H), 4.91 (s, 2H), 4.19 (m, IH), 0.85 (d, J = 6.4 Hz, 3H). Tert-butyl ( 1 R,2S)- 1 -(4H-benzord1 Γ 1 ,31dioxin-7-yl)- 1 -hvdroxypropan-2-ylcarbamate (I3b)

APCI-MS: m/z 210 [MH+ -BOC]
1H NMR (400 MHz, DMSO- ¾ δ 6.97 (1H, d), 6.88 (1H, d), 6.77 (1H, s), 6.56 (1H, d), 5.27 (1H, d), 5.22 (2H, s), 4.83 (2H, s), 4.44 (1H, t), 3.53 (1H, m), 1.32 (9H, s), 0.93 (3H, d). (S)-Tert-butyl 1 -(4H-benzord1 Γ 1 ,31dioxin-7-vD- 1 -oxopropan-2-ylcarbamate (I3c)

In a separate reaction tube (S)-tert-butyl l-(methoxy(methyl)amino)-l-oxopropan-2- ylcarbamate (1 g, 4.31 mmol) was suspended in THF (5 mL) and cooled in an ice/acetone bath to below -5 °C. Isopropylmagnesium chloride, 2M solution in THF (2.5 mL, 5.00 mmol) was slowly added to form a solution. To this solution was added the above freshly prepared Grignard reagent. The mixture was allowed to reach r.t. and stirred for 4 hours. The reaction mixture was slowly poured into ice-cold 150 mL 1M HC1. Ethyl acetate (150 mL) was added and the mixture was stirred for a few minutes and transferred to a separation funnel. The organic phase was washed with water and brine, dried over MgS04, filtered and concentrated. The obtained crude product was further purified by flash chromatography using a prepacked 70g silica column with a gradient of 10% TBME to 40% TBME in heptane as eluent. The subtitle compound was obtained as a colourless solid. Yield 790 mg (59%>)
APCI-MS: m/z 208 [MH+ -BOC]
1H NMR (400 MHz, DMSO-^) δ 7.53 (IH, dd), 7.39 (IH, s), 7.30 (IH, d), 7.22 (IH, d), 5.30 (2H, s), 4.98 (IH, m), 4.95 (2H, s), 1.35 (9H, s), 1.20 (3H, d).
Preparation 4
3-(5-([(lR,2S)-2-[(2,2-difluoropropanoyl)aminol-l-(2,3-dihydro-l,4-benzodioxin-6- yl)propyl]oxy| - 1 H-indazol- 1 -yl)-N-[(3R)-tetrahydrofuran-3-yllbenzamide

APCI-MS: m/z 607 [MH+]
1H NMR (400 MHz, DMSO-d6) δ 8.71 (IH, d), 8.65 (IH, d), 8.24 (IH, s), 8.18 (IH, s), 7.90 - 7.84 (2H, m), 7.77 (IH, d), 7.65 (IH, t), 7.21 (IH, dd), 7.13 (IH, d), 6.89 - 6.78 (3H, m), 5.17 (IH, d), 4.48 (IH, m), 4.23 - 4.10 (5H, m), 3.89 - 3.82 (2H, m), 3.72 (IH, td), 3.61 (IH, dd), 2.16 (IH, m), 1.94 (IH, m), 1.55 (3H, t), 1.29 (3H, d).
LC (method A) rt = 12.03 min
LC (method B) rt = 11.13 min
Chiral SFC (method B) rt = 4.71 min
M.p. = 177 °C
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015080434 | 2015-03-19 | PHENYL AND BENZODIOXINYL SUBSTITUTED INDAZOLES DERIVATIVES |
| US8916600 | 2014-12-23 | Phenyl and benzodioxinyl substituted indazoles derivatives |
| US8211930 | 2012-07-03 | Phenyl and Benzodioxinyl Substituted Indazoles Derivatives |
https://www.astrazeneca.com/content/dam/az/press-releases/2014/Q2/Pipeline-table.pdf
////////AZD 7594, AZ13189620, AZD-7594 , phase 2, astrazeneca, 1196509-60-0
c21cc(ccc1n(nc2)c3cc(ccc3)C(=O)NC4COCC4)O[C@H](c5cc6c(cc5)OCCO6)[C@@H](NC(=O)C(F)(F)C)C
CC(C(C1=CC2=C(C=C1)OCCO2)OC3=CC4=C(C=C3)N(N=C4)C5=CC=CC(=C5)C(=O)NC6CCOC6)NC(=O)C(C)(F)F
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
P.STHE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP







 .
.