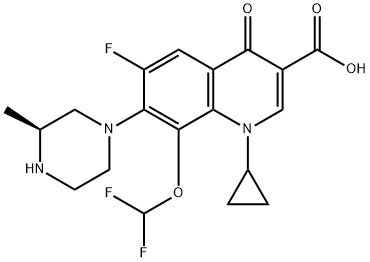
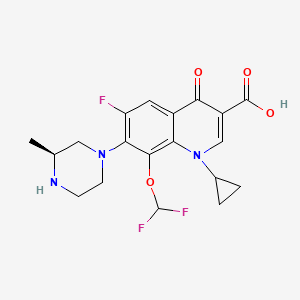
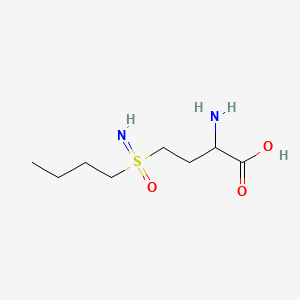
Buthionine Sulphoximine
NDA Filed in china
A gamma-glutamylcysteine synthetase inhibitor potentially for the treatment of solid tumors.
NSC-326231; BSO
CAS No. 5072-26-4
BUTHIONINE SULFOXIMINE; DL-Buthionine-[S,R]-sulfoximine; 5072-26-4; Buthionine sulfoxamine; Buthionine-S,R-sulfoximine; Buthione sulfoximine;
| Molecular Formula: | C8H18N2O3S |
|---|---|
| Molecular Weight: | 222.30512 g/mol |
Buthionine sulfoximine (BSO) is a sulfoximine which reduces levels of glutathione and is being investigated as an adjunct withchemotherapy in the treatment of cancer.[1] The compound inhibits gamma-glutamylcysteine synthetase, the enzyme required in the first step of glutathione synthesis. Buthionine sulfoximine may also be used to increase the sensitivity of parasites to oxidativeantiparasitic drugs.[2]
Buthionine sulphoximine is an oncolytic agent in early clinical development at the National Cancer Institute (NCI) for the treatment of neuroblastoma in pediatric patients in combination with melphalan and bone marrow or peripheral stem cell transplantation.
DATA

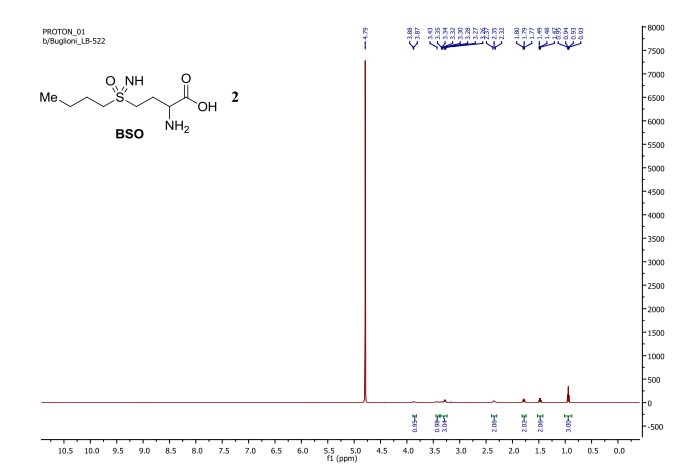
1H NMR
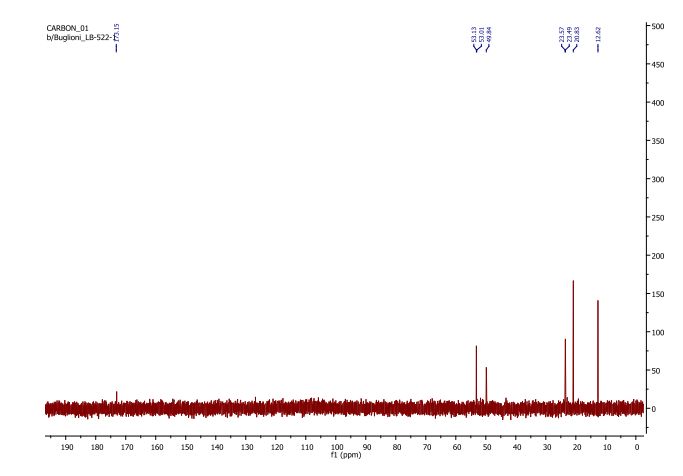
13C NMR
Synthesis
Methionine and buthionine sulfoximines: Syntheses under mild and safe imidation/oxidation conditions
Advanced Synthesis&Catalysis (2014), 356, (10), 2209-2213
Advanced Synthesis&Catalysis (2014), 356, (10), 2209-2213
Abstract

Methionine and buthionine sulfoximines (MSO and BSO) are non-natural amino acids known to inhibit the biosynthesis of glutathione (GSH). The current syntheses of these biologically active molecules involve harsh reaction conditions and the use of hazardous reagents for the sulfur imidation. Here, improved syntheses of MSO and BSO are presented including safe and mild one-pot imidation/oxidation sequences and single-step deprotections of three different functionalities.
Methionine and Buthionine Sulfoximines: Syntheses under Mild and Safe Imidation/Oxidation Conditions
DOI: 10.1002/adsc.201400354
http://onlinelibrary.wiley.com/doi/10.1002/adsc.201400354/abstractReferences
- Defty, CL; Marsden, JR (2012). "Melphalan in regional chemotherapy for locally recurrent metastatic melanoma.". Current topics in medicinal chemistry 12 (1): 53–60. PMID 22196271.
- "Definition of buthionine sulfoximine - National Cancer Institute Drug Dictionary".
 | |
 | |
| Names | |
|---|---|
| IUPAC name
2-amino-4-(butylsulfonimidoyl)butanoic acid
| |
| Other names
BSO
| |
| Identifiers | |
| CAS Number | 5072-26-4 |
| ChEBI | CHEBI:28714 |
| ChemSpider | 19896 |
| Jmol 3D model | Interactive image |
| MeSH | Buthionine+sulfoximine |
| PubChem | 21157 |
| Properties | |
| Chemical formula | C8H18N2O3S |
| Molar mass | 222.305 g/mol |
| Density | 1.29 g/mL |
| Melting point | 215 °C (419 °F; 488 K) |
| Boiling point | 382.3 °C (720.1 °F; 655.5 K) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
CCCCS(=N)(=O)CCC(C(=O)O)N











.bmp)
.bmp)
.gif)