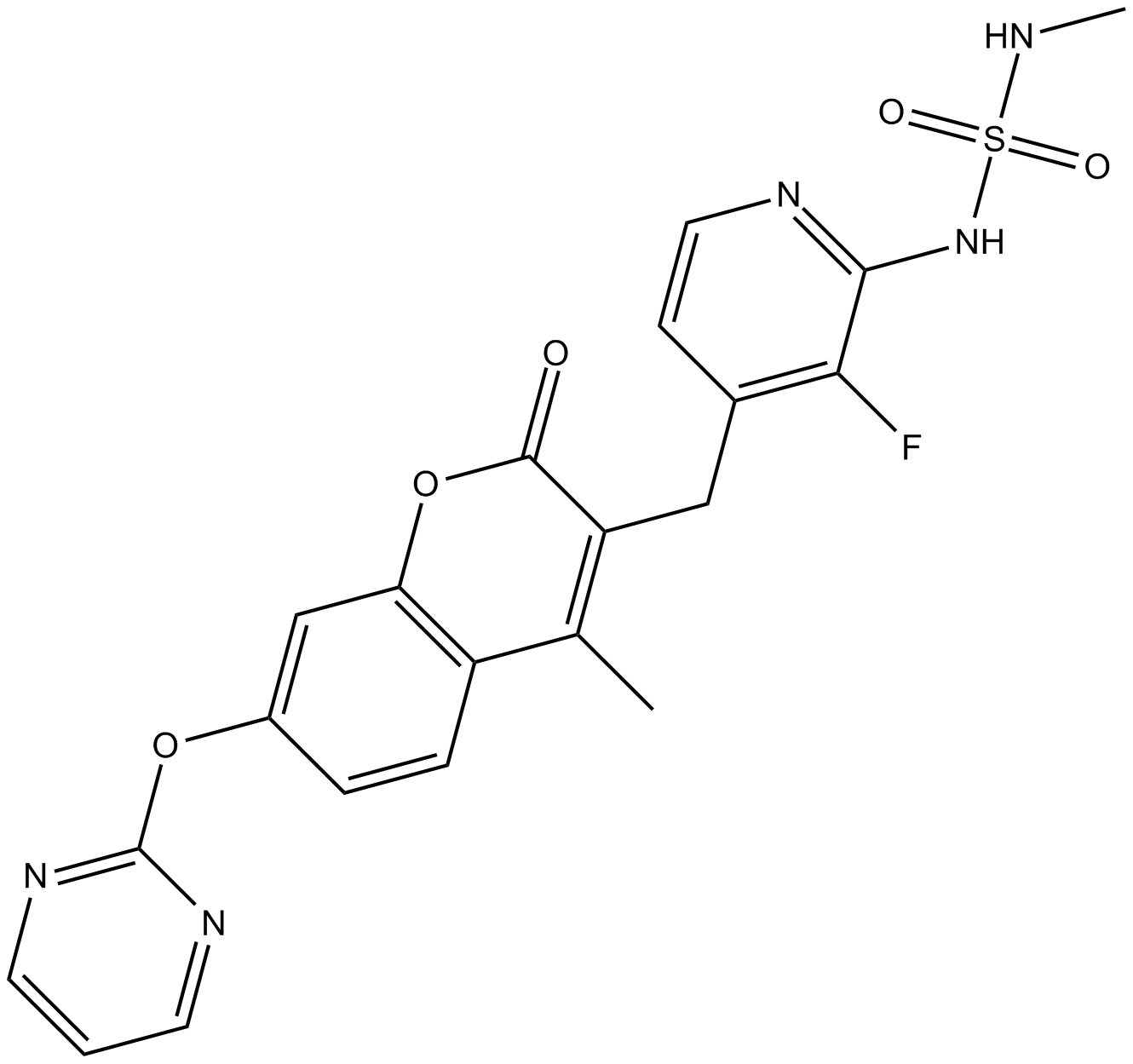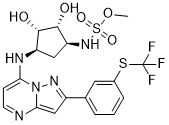
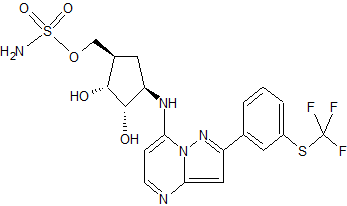
TAK-243, AOB 87172, MLN-7243
CAS 1450833-55-2
Chemical Formula: C19H20F3N5O5S2
Molecular Weight: 519.5142
Chemical Formula: C19H20F3N5O5S2
Molecular Weight: 519.5142
Sulfamic acid, [(1R,2R,3S,4R)-2,3-dihydroxy-4-[[2-[3-[(trifluoromethyl)thio]phenyl]pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]methyl ester
((lR,2R,3S,4R)-2,3-dihydroxy-4-(2-(3-(trifluoromethylthio)phenyl)pyrazolo[l
,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamate
methyl
((1S,2R,3S,4R)-2,3-dihydroxy-4-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclopentyl)sulfamate
Phase I
Millennium Pharmaceuticals, Inc. INNOVATOR
Roushan AFROZE, Indu T. Bharathan,Jeffrey P. CIAVARRI, Paul E. Fleming,Jeffrey L. Gaulin, Mario Girard, Steven P. Langston, Francois R. SOUCY, Tzu-Tshin WONG, Yingchun Ye,
A UAE inhibitor potentially for the treatment of solid tumors.TAK-243, also known as MLN7243 and AOB87172, is a small molecule inhibitor of ubiquitin-activating enzyme (UAE), with potential antineoplastic activity. UAE inhibitor MLN7243 binds to and inhibits UAE, which prevents both protein ubiquitination and subsequent protein degradation by the proteasome. This results in an excess of proteins in the cells and may lead to endoplasmic reticulum (ER) stress-mediated apoptosis. This inhibits tumor cell proliferation and survival. UAE, also called ubiquitin E1 enzyme (UBA1; E1), is more active in cancer cells than in normal, healthy cells.
Research Code TAK-243; MLN-7243, TAK-243; TAK 243; TAK243; MLN7243; MLN-7243; MLN 7243; AOB87172; AOB-87172; AOB 87172.
CAS No. 1450833-55-2(MLN 7243)
- Originator Millennium
- Developer Takeda Oncology
- Class Antineoplastics
- Mechanism of Action Ubiquitin-protein ligase inhibitors
- Phase I Solid tumours

Most Recent Events
- 01 Feb 2014 Phase-I clinical trials in Solid tumours (late-stage disease, second-line therapy or greater) in USA (IV)
- 18 Dec 2013 Preclinical trials in Solid tumours in USA (IV)
- 18 Dec 2013 Millennium plans a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA (NCT02045095)
Cancer is the second most common cause of death in the U.S. and accounts for one of every eight deaths globally (American Cancer Society, Cancer Facts and Figures, 2014). The American Cancer Society expects that in 2014 at least 1,665,540 new cancer cases will be diagnosed in the US and 585,720 Americans are expected to die of cancer, almost 1 ,600 people per day. Currently available paradigms for treating solid tumors may include systemic treatment such as chemotherapy, hormonal therapy, use of targeted agents and biological agents, either as single agents or in combination. These treatments can be delivered in combination with localized treatments such as surgery or radiotherapy. These anti-cancer paradigms can be use in the curative setting as adjuvant or neo-adjuvant treatments or in the metastatic setting as palliative case for prolonged survival and to help manage symptoms and side-effects. In hematological cancers, stem cell transplants may also be an option in certain cancers as well as chemotherapy and/or radiation. Although medical advances have improved cancer survival rates, there remains a continuing need for new and more effective treatments.
Ubiquitin is a small 76-amino acid protein that is the founding member of a family of posttranslational modifiers known as the ubiquitin-like proteins (Ubls). Ubls play key roles in controlling many biological processes including cell division, cell signaling and the immune response. There are 8 known human Ubl activating enzymes (known as Els) (Schulman, B.A., and J.W. Harper, 2009, Ubiquitin-like protein activation by El enzymes: the apex for downstream signalling pathways, Nat Rev Mol Cell Biol 10:319-331). Ubiquitin and other Ubls are activated by a specific El enzyme which catalyzes the formation of an acyl-adenylate intermediate with the C-terminal glycine of the Ubl. The activated Ubl molecule is then transferred to the catalytic cysteine residue within the El enzyme through formation of a thioester bond intermediate. The El -Ubl intermediate and an E2 enzyme interact, resulting in a thioester exchange wherein the Ubl is transferred from the El to active site cysteine on the E2. The Ubl is then conjugated, i.e. transferred, to the target protein, either directly or in conjunction with an E3 ligase enzyme, through isopeptide bond formation with the amino group of a lysine side chain in the target protein. Eukaryotic cells possess ~35 ubiquitin E2 enzymes and >500 ubiquitin E3 enzymes. The E3 enzymes are the specificity factors of the ubiquitin pathway which mediate the selective targeting of specific cellular substrate proteins (Deshaies, R.J., and C.A. Joazeiro, 2009, RING domain E3 ubiquitin ligases, Annu Rev Biochem 78:399-434; Lipkowitz, S., and A.M. Weissman, 2011, RTNGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis, Nat Rev Cancer 11 :629-643; Rotin, D., and S. Kumar, 2009, Physiological functions of the HECT family of ubiquitin ligases, Nat Rev Mol Cell Biol 10:398-409).
Two El enzymes have been identified for ubiquitin, UAE (ubiquitin-activating enzyme) and UBA6 (Jin, J., et al., 2007, Dual El activation systems for ubiquitin differentially regulate E2 enzyme charging, Nature 447: 1135-1138). UAE is the El responsible for the majority (>99%) of ubiquitin flux within the cell. UAE is capable of charging each of the approximately -35 E2 enzymes with the exception of Usel, which is the only E2 known to exclusively work with UBA6 (Jin et al., 2007). Inhibition of UAE is sufficient to dramatically impair the great majority of ubiquitin-dependent cellular processes (Ciechanover, A., et al., 1984, Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85, Cell 37:57-66; Finley, D., A. et al., 1984, Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85, Cell 37:43-55).
The cellular signals generated by ubiquitin are diverse. Ubiquitin can be attached to substrates as a single entity or as polyubiquitin polymers generated through isopeptide linkages between the C-terminus of one ubiquitin and one of the many lysines on a second ubiquitin. These varied modifications are translated into a variety of cellular signals. For example, conjugation of a lysine 48 -linked polyubiquitin chain to a substrate protein is predominantly associated with targeting the protein for removal by the 26S proteasome. A single ubiquitin modification, or monoubiquination, typically affects protein localization and/or function. For example, monoubiquitination modulates the following: 1) the function of Histones 2a and 2b (Chandrasekharan, M.B., et al., 2010, Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation, Epigenetics 5:460-468), 2) controls the nucleo-cytoplasmic shuttling of PTEN (Trotman, L,C, et al., 2007, 3) ubiquitination regulates PTEN nuclear import and tumor suppression, Cell 128: 141-156), 4) drives localization of the FANCD2 protein to sites of DNA damage (Gregory, R.C., et al., 2003, Regulation of the Fanconi anemia pathway by monoubiquitination, Semin Cancer Biol 13:77-82) and 5) promotes the internalization and endosomal/lysosomal turnover of some cell surface receptors, like EGFR (Mosesson, Y., and Y. Yarden, 2006, Monoubiquitylation: a recurrent theme in membrane proteintransport. Isr Med Assoc J 8:233-237). Other forms of polyubiquitination chains occur on lysine positions 11, 29 and 63, impacting various cellular roles including cell cycle, DNA repair and autophagy (Behrends, C, and J.W. Harper, 2011, Constructing and decoding unconventional ubiquitin chains, Nat Struct Mol Biol 18:520-528; Bennett, E.J., and J.W. Harper, 2008, DNA damage: ubiquitin marks the spot, Nat Struct Mol Biol 15:20-22; Komander, D., 2009, The emerging complexity of protein ubiquitination, Biochem Soc Trans 37:937-953).
UAE-initiated ubiquitin conjugation plays an important role in protein homeostasis, cell surface receptor trafficking, transcription factor turnover and cell cycle progression. Many of these processes are important for cancer cell survival and it is believed that tumor cells may have increased sensitivity to UAE inhibition as a result of their rapid growth rate, increased metabolic demands and oncogene fueled protein stress. Preclinical studies with PYZD-4409, a UAE inhibitor, demonstrated this compound induced cell death in both leukemia and myeloma cell lines and induced anti-tumor activity in a mouse acute myeloid leukemia (AML model). (Xu, W.G., et al., 2010, The ubiquitin-activating enzyme El as a therapeutic target for the treatment of leukemia and multipie myeloma, Blood, 115:2251-59). Thus, UAE represents a protein homeostasis target opportunity for the treatment of cancer.
Abstracts: AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; November 5-9, 2015; Boston, MA
Abstract
Clinical
results of VELCADE® (bortezomib) For Injection have prompted evaluation
of other enzymes within the ubiquitin proteasome system (UPS) as
druggable targets for human cancer. We have identified a first in class
investigational drug, TAK-243 (MLN7243), which targets the ubiquitin
activating enzyme, UAE (UBA1), an essential cellular enzyme responsible
for activating > 99% of all cellular ubiquitin. Ubiquitin is involved
in multiple cellular processes including ubiquitin-dependent protein
turnover, cell cycle progression, regulation of apoptosis, protein
localization and response to DNA damage. Experiments combining targeted
siRNA knockdown with TAK-243 identified DNA damage repair genes
necessary for UAE inhibitor-induced cell death. A more focused approach
revealed TAK-243 treatment blocked essential monoubiquitination events
within the Translesion synthesis (TLS), Fanconi Anemia (FA) and
Homologous recombination (HR) pathways. Inhibition of UAE prevented
mono-ubiquitin signaling of key mediators within these pathways,
including PCNA and FANCD2, by blocking formation of their specific
E2-ubiquitin thioesters. In vitro cell-based assays combining
TAK-243 with ultraviolet (UV) and radiation, both known to induce DNA
damage, yielded inhibition of cell growth and enhanced DNA damage as
observed through colony formation assays and Comet assay detection,
respectively. Xenograft tumor bearing mice were treated with carboplatin
or docetaxel, combined with TAK-243, to evaluate combination benefits in vivo.
Synergistic and additive anti-tumor combination benefits were observed
in animals treated with TAK-243 + carboplatin and TAK-243 + docetaxel.
These important mechanistic in vitro and in vivo
studies indicate the dependency of ubiquitination signaling in DNA
damage repair and provide a mechanistic rationale for combining
radiation, carboplatin or docetaxel with TAK-243 in the clinical
setting. Currently, TAK-243 is being evaluated in a solid tumor phase I
clinical trial evaluating safety, tolerability, pharmacokinetics,
pharmacodynamics and anti-tumor activity (ClinicalTrials.gov identifier:
NCT02045095).
Citation Format:
Michael A. Milhollen, Judi Shi, Tary Traore, Jessica Huck, Darshan
Sappal, Jennifer Duffy, Eric Lightcap, Yuko Ishii, Jeff Ciavarri, Paul
Fleming, Neil Bence, Marc L. Hyer. The small molecule UAE inhibitor
TAK-243 (MLN7243) prevents DNA damage repair and reduces cell
viability/tumor growth when combined with radiation, carboplatin and
docetaxel. [abstract]. In: Proceedings of the AACR-NCI-EORTC
International Conference: Molecular Targets and Cancer Therapeutics;
2015 Nov 5-9; Boston, MA. Philadelphia (PA): AACR; Mol Cancer Ther
2015;14(12 Suppl 2):Abstract nr A164.
PATENT
WO 2013123169
https://www.google.com/patents/WO2013123169A1?cl=en
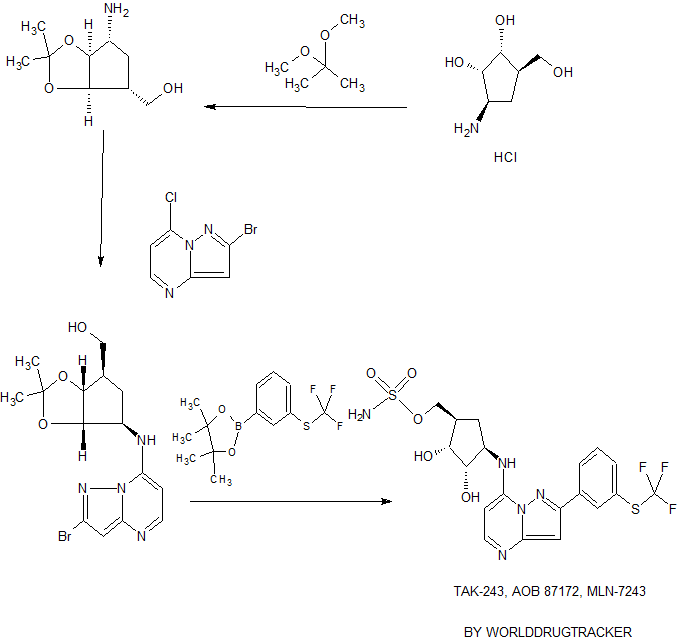
Scheme 1 : General route for 2-substituted ((1R,2R,3S,4R)-2,3-dihydroxy-4- (pyrazolo[1,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates

Scheme 2: General route for 5-halogen substituted, 2 -substituted ((1R,2R,3S,4R)- 2,3-dihydroxy-4-(pyrazolo[1,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates


Scheme 3: General route for 5-alkyl substituted, 2-substituted ((1R,2R,3S,4R)-2,3- dihydroxy-4-(pyrazolo[1 ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates

Example 17. Synthesis of (s.e.)-{(1 ,2R,3S,4R)-4-[(3,6-dichloro-2-{3- [(trifluoromethyl)sulfanyl]phenyl}pyrazolo[1,5-a]pyrimidin-7-yl)amino]-2,3- dihydroxycyclopentyl}methyl sulfamate (1-124) and (s.e.)-{(1 ,2R,3S,4R)-4-[(6-chloro-2-{3- [(trifluoromethyl)sulfanyl]phenyl}pyrazolo[1,5^]pyrimidin-7-y[)arnino]-2,3- dihydroxycyclopentyl}methyl sulfamate 0-125).

SIMILAR NOT SAME
Step 1. To a vial containing s.e {(1 ,2 ,3S,4 )-2,3-dihydroxy-4-t(2-{3- [(t rif I u orometh y l)sulf a nyl] phen l}p^sulfamate (0.82 g, 0.0015 mol) and cooled to 0 °C is added N-chlorosuccinimide (126 mg, 0.000943 mol) as a solution in 12 mL of N,N-dimethy)formamide. The reaction mixture is stirred overnight with warming to rt. Saturated sodium bicarbonate solution is added and the reaction mixture is extracted with ethyl acetate, washed with brine, dried over sodium sulfate and concentrated in vacuo. The crude material is first purified by column chromatography (eluent: methanol/methylene chloride) and then purified by HPLC to afford both the dichloro (LCMS: (FA) +1 588) and mono chloro (LCMS: (FA) M+1 554) titlecompounds.
PATENT
WO 2016069393
UAE inhibitors are disclosed in patent application publications WO2013/123169 and US 2014/0088096. In one embodiment, the UAE inhibitor is a compound having the following structure (Compound 1):

(Compound 1);
or a pharmaceutically acceptable salt thereof. The Compound 1 is named ((lR,2R,3S,4R)-2,3-dihydroxy-4-(2-(3-(trifluoromethylthio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamate.
process for making Compound 1 :

Compound 1;
or pharmaceutically acceptable salt thereof, comprising the steps of:
a) contacting Compound 9 or a salt, solvate or hydrate thereof with 2,2-dimethyl-l,3-dioxane-4,6-dione (Meldrum's acid):

Compound 9
under coupling conditions to provide compound 8 or a salt, solvate or hydrate thereof:

Compound 8
b) subjecting compound 8 or a salt, solvate or hydrate thereof to cyclization conditions to provide compound 7 or a salt, solvate or hydrate thereof

Compound 7
c) contacting Compound 7 or a salt, solvate or hydrate thereof with benzotriazole under chlorination displacement conditions to provide Compound 5 or a salt, complex, solvate or hydratei thereof

; Compound 5
d) contacting Compound 5 or a salt, complex, solvate or hydrate thereof with Compound 6 or a solvate or hydrate thereof:

; Compound 6
under displacement reaction conditions to provide Compound 3 or a salt, solvate or hydrate thereof
solvate or hydrate thereof with Compound

Cl ; Compound 4
under sulfamoylating reaction conditions to provide Compound 2 or a salt, solvate or hydrate thereof

; Compound 2
f) contacting Compound 2 or a salt, solvate or hydrate thereof with an acid under sulfamoylation conditions to provide Compound 1 or a pharmaceutically acceptable salt thereof
 COMPD1
COMPD1Example 1: Synthesis of S-iB-Ktrifluoromethyltsulfanyllphenyll-lH-pyrazol-S-amine
Step A: 3-((trifluoromethyl)thio)benzoate
To dimethylcarbonate (68 mL) was added 3-((trifluoromethyl)thio)benzoic acid (100 g, Beta Pharma Scientific) and a catalytic amount of sulfuric acid (2.4 mL). The mixture was then heated to 90°C for 5h. The reaction was then cooled to room temperature and quenched with sodium bicarbonate (1.0 L). To the aqueous layer was with ethyl acetate (1.0 L). The phases were separated and this process was repeated with ethyl acetate (1.0 L). The organic layers were combined and concentrated with a rotovap to give a light orange oil. The methyl 3-((trifluoromethyl)thio)benzoate (105g, 99%) was taken on crude to the next reaction. Ή NMR (300 MHz, CHLOROFORM-^ δ ppm 3.99 (s, 3 H) 7.49 - 7.58 (m, 1 H) 7.85 (d, J=l.62 Hz, 1 H) 8.17 (dt, J=7.69, 1.43 Hz, 1 H) 8.32 - 8.44 (m, 1 H).
Step B: 3-oxo-3-(3-((trifluoromcthvnthio)phcnyl>proDaneiiitrilc
Methyl 3-((trifluoromethyl)thio)benzoate (100.0 g) in tetrahydrofuran (1.0 L) was added acetonitrile (44.2 mL, 847 rnmol) and 1M (in THF) potassium tert-butoxide (95.01 g). The reaction was complete in 10 min by HPLC analysis. The reaction was quenched with 1M HC1 (1.0 L) and then extracted with three times with (1.0 L) of ethyl acetate. The organic layers with 3-oxo-3-(3-((trifluoromethyl)thio)phenyl)propanenitrile were then concentrated to dryness. This material (lOO.Og, 96.3%) was taken on crude with further purification. Ή NMR (300 MHz, CHLOROFORM-rf) δ ppm 4.12 (s, 2 H) 7.51 - 7.75 (m, 1 H) 7.89 - 8.01 (m, 1 H) 8.01 - 8.10 (m, 1 H) 8.20 (s, 1 H)
Step C: 3-}3-htrifliioromethv sulfan llphenyl}-lH-pyrazol-5-amine
[0152] To 3-oxo-3-{3-[(trifluoromethyl)sulfanyl]phenyl}propanenitrile (100.0 g,) in ethanol (1000.0 mL) was added hydrazine hydrate (59.52 mL). The reaction was heated to 100°C for lh at which point HPLC analysis showed the reaction was complete. The reaction was concentrated to dryness on a rotovap to give a brown oil. The oil was taken up in ethyl acetate (1.0 L) and extracted with water (1.0 L). The phases were separated and the organic phase was concentrated. Upon concentration 3-{3-[(trifluoromethyl)sulfanyl]phenyl}-lH-pyrazol-5-amine was obtained (80.8 g; Yield = 76.4%) . !H NMR (300 MHz, CHLOROFORM-^ δ ppm 5.95 (s, 1 H) 6.73 (br s, 1 H) 7.13 - 7.34 (m, 2 H) 7.42 - 7.74 (m, 3 H) 7.85 (s, 1 H).
Example 2: f R.2R.3St4RV2.3-dihvdroxy-4-ff2-r3- ((trifluoromethylHhio)phenvnpyrazolo[l,5-alpyrimidin-7-yl¼mino)cvclopentyl)metliyl sulfamate
Step 1: f2.2-dimethyl-5-ffl3-(3-((triiluoromethvnthio phenvn-lH-pyrazol-5- amino methyleBC>-1.3-dioxane-4,6-dione)
[0155] To trimethoxy orthoformate (2.0 L), at 20°C and under a blanket of nitrogen, was added 2,2-dimethyl-l,3-dioxane-4,6-dione (361.35 g). The resulting white suspension went clear within minutes and was heated to 85°C over 15 minutes. The reaction was held at 85°C for 120 minutes. While the reaction was heated and stirred another solution of 3-(3-((trifluoromethyl)thio)pheny])-lH-pyrazol-5-amine (500.0 g) was made. To a 4L RBF was added 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (500.0 g) and then trimethoxy orthoformate (1.4 L) added into this solid. This solution was mixed to dissolve the solids and resulted a dark brown solution. The solution of 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (-1.8L in trimethoxy orthoformate) was added to the reactor over 30 minutes while maintaining the reaction temperature at 85°C. The reaction was then stirred for 20 minutes with white solids forming in the solution. After 20 minutes the reaction was sampled and the UPLC showed the complete conversion of 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5 -amine to 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l ,3-dioxane-4,6-dione. The reaction was cooled to 20 °C over 20 minutes and maintained at that temperature for 20 additional minutes. At this point, a thick white slurry had formed and the reaction was filtered using a Nutche Filter over 15 minutes. The reactor was washed with 1L of ethyl acetate and this solution was then mixed with the filter cake and removed by filtration. The cake was dried for -40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16 hours). The reaction was then analyzed by FfPLC and NMR to give 2,2-dimethyl-5-(((3-(3 -((trifluoromethyl)thio)phenyi lH-pyrazol-5-yl)amino)methylene)- 1 ,3-dioxane-4,6-dione (635.3 g, 79%) XH NMR (300 MHz, DMSO-cfe) δ ppm 1.68 (s, 6 H) 7.05 (d, J=2.05 Hz, 1 H) 7.64 -7.77 (m, 2 H) 7.77 - 8.03 (m, 1 H) 8.12 (s, 1 H) 8.72 (d, J=14.36 Hz, 1 H) 1 1.35 (d, J=14.66 Hz, 1 H) 13.47 (s, 1 H).
[0156] Step 2: 2-( 3-f(trifluoromethyl)thio phenyl)pyrazoIo [1,5-al pyrimidin-7-ol
[0157] A solution of 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l,3-dioxane-4,6-dione (615.00 g) in 1,2-dichIorobenzene (6.3 L) was stirred at ambient temperature for 10 minutes. The solution was then heated to 150°C over 75 minutes. The reaction was maintained at this temperature for 16 hours. An sample was taken after 16 hours and the UPLC analysis showed the complete conversion of 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yI)amino)methylene)-l,3-dioxane-4,6-dione to 2-(3- ((trifluoromethyl)tmo)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol. The reaction was cooled to 20°C over 130 minutes. At this point, a thick white slurry had formed and the reaction was filtered using a Nutche Filter over 15 minutes. The reactor was washed with 1.8 L of acetonitrile and this solution was then mixed with the filter cake and then the solvent was removed by filtration. The cake was dried for ~40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16 hours). The reaction was then analyzed by HPLC and NMR to give 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (331.2 g, 72%) Ή NMR (300 MHz, METHANOL-^) δ ppm 6.55 (d, J=7.33 Hz, 1 H) 7.59 (s, 1 H) 8.40 - 8.52 (m, 1 H) 8.53 - 8.64 (m, 1 H) 8.69 (d, J=7.62 Hz, 1 H) 9.01 (dt, J=7.77, 1.39 Hz, 1 H) 9.12 (s, 1 H) 13.34 (s, 1 H).
[0158] Step 3: l-(2-(3-(f trffluoromethvmhiotohenvnpyrazolo n.5-al pyrimidin-7-vn-lH-benzofdiri.2.31triazole: triethylamine: hydrochloride complex (1:1.25:1.25 molesimolestmolest
[0159] To a solution of 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (30.00 g), benzotriazole (287.02 g) in acetonitrile (3000 mL) and triethylamine (403.00 mL) at 0°C, was added phosphoryl chloride (108 mL) slowly under a blanket of nitrogen, maintaining < 10°C. The reaction was then warmed to 80°C over 45 minutes and stirred for 240 minutes. HPLC indicated complete
consumption of starting material. To the reaction mixture was added acetonitrile (3000 mL) while maintaining the temperature at 80°C. The reaction was then cooled to 20°C over 80 minutes. The reaction was then stirred at ambient temperature for 14 hours. At this point, a thick slurry had formed and the reaction was filtered using a Nutche filter over 15 minutes. The reactor was washed twice with 900 mL of acetonitrile and this solution was then mixed with the filter cake and then the solvent was removed by filtration. The cake was dried for -40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16h). The reaction was then analyzed by HPLC and NMR to give l-(2-(3-((trifluorometJiyl)thio)phe
triethylamine: hydrochloride complex (1:1.25:1.25 moles:moles:moles) (438.1 g, 83%). ¾ NMR (300 MHz, DMSO-</6) δ ppm 1.19 (t, J=7.33 Hz, 12 H) 3.07 (qd, J=7.28, 4.84 Hz, 8 H) 7.60 - 7.78 (m, 6 H) 7.80 - 7.87 (m, 1 H) 8.15 (dt, J=7.99, 1.28 Hz, 1 H) 8.24 (s, 1 H) 8.33 (dt, J=8.14, 0.92 Hz, 1 H) 8.85 (d, J=4.69 Hz, 1 H).
[0160] Step 4: ff3aR4R.6R.6aS 2.2-dimethyl-6-ff2-f3~mrifluoromethyl)thio)phenvnpyrazoloil.5-alD\timidin-7-yl¼mino)tctralivdro-3aH-cvcLoDentaldlll,31dioxol-4-vnincthanol
[0161] To the reactor was added l-(2 3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)-lH-benzo[d][l,2,3]triazole: triethylamine: hydrochloride complex (1 :1.25: 1.25 moles :moles:moles) (430.0 g) and ((3aR,4R,6R,6aS)-6-amino-2,2-dimethyltetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol hydrochloride (209.0 g) and then triethylamine (2103 mL) was added. The reaction was then heated to 80°C, under a blanket of nitrogen. After 360 minutes, HPLC analysis indicated that the reaction mixture contained <1% starting material and the reaction was cooled to 20°C over 60 minutes. To the reaction was added ethyl acetate (3.5 L) and water (3.5 L). After stirring for 10 minutes the phases were separated and the aqueous layer was back extracted with ethyl acetate (3.5 L). The organic layers were combined and concentrated to form a dark, brown oil. Acetonitrile (4.5 L) was added and the solution was concentrated to dryness to give an orange solid. The solids was transferred back to the reaction with water (4.3 L), heated to 50°C, and stirred for 20 minutes. White solids formed in this hot solution and were isolated by filtration using a Nutche Filter over 15 minutes. The solids were dried under vacuum for 15 minutes on the filter and then dissolved in acetonitrile (4.0 L) at 0°C. The solution was stirred for 1 minutes. The solution was then filtered through a fritted funnel to remove the hydrolysis solid by product and the solution was concentrated to dryness. The solids were dried in a vacuum oven at full vacuum overnight (40°C, 16 hours). The reaction was then analyzed by HPLC and NMR to give ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5 -a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanoI (349.2 g, 88%). Ή NMR (300 MHz, DMSO-<¾) δ ppm 1.25 (s, 3 H) 1.47 (s, 3 H) 1.76 - 1.90 (m, 1 H) 2.25 (br d, J-3.22 Hz, 1 H) 2.33 - 2.47 (m, 1 H) 3.46 - 3.67 (m, 2 H) 4.08 (br d, J=5.57 Hz, 1 H) 4.48 - 4.64 (m, 2 H) 5.19 (t, J=4.40 Hz, 1 H) 6.28 (d, J=5.28 Hz, 1 H) 7.06 (s, 1 H) 7.58 - 7.71 (m, 1 H) 7.72 - 7.80 (m, 1 H) 8.12 - 8.24 (m, 2 H) 8.31 (d, J=7.62 Hz, 1 H) 8.42 (s, 1 H).
[0162] Step 5: ((3aR.4R.6R.6aS 2.2-dimethyl-6-ff2-f3-fftrifluoroinethYmhio)phenvnpyrazolo[1.5-al Dyrimidin-7-vnan] iiio>tetrahvdro-3aH-cvclonen ta [dl [1,31 dioTOl-4-yl )meth yl tert-bntoxycarbonylsulfamate
[0163] ((3aR,4R,6R,6aS)-2,2-dime l-6-((2-(3-((trifluorome
7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol (6.0 g) was dissolved in 2-methyltetrahedrafuran (60.0 mL) and to this solution was added pyridinium p-toluenesulfonate (5.9 g). This formed a precipitated and to this white slurry was added (4-aza-l-azoniabicyclo[2.2.2]oct-l-ylsulfonyl)(tert-butoxycarbonyl)azanide-l,4-diazabicyclo[2.2.2]octane (1 :1) hydrochloride1 (17.0 g). The mixture was stirred at ambient temperature until the HPLC showed <1% ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol remaining starting material (-300 minutes). The reaction was quenched with water (60 mL) and the phases were separated. To the organic layer was added acetonitrile (60 mL) and the mixture was concentrated using a rotovap at 50°C to ~60 mL. The mixture was allowed to cool to room temperature and stirred overnight. During this time a white slurry formed. White solids were filtered using a medium fritted filter. The solid was dried in a vacuum oven at full vacuum overnight (40 °C). The reaction was then analyzed by HPLC and NMR to give ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyI)tM^
cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (5.03 g, 68%). [H NMR (300 MHz, DMSO- 6) δ ppm 1.26 (s, 3 H) 1.42 (s, 9 H) 1.51 (s, 3 H) 2.33 - 2.48 (m, 2 H) 3.30 (br s, 1 H) 4.06 - 4.21 (m, 1 H) 4.29 (d, J=5.28 Hz, 2 H) 4.52 (dd, J=7.18, 5.13 Hz, 1 H) 4.76 (dd, J=7.18, 4.54 Hz, 1 H) 6.35 (d, J=5.57 Hz, 1 H) 7.08 (s, 1 H) 7.63 - 7.72 (m, 1 H) 7.74 - 7.82 (m, 1 H) 8.01 (d, ^=7. 2 Hz, 1 H) 8.21 (d, J=5.28 Hz, 1 H) 8.31 (dt, J=7.84, 1.36 Hz, 1 H) 8.48 (s, 1 H) 1 1.92 (br s, 1 H)
[0164] Step 6: f R,2R3S.4R)-2J-dihvdroxy-4-((2-(3-fftrifluoromethvDthio^phenvnpyrazolori.5-a]pyrimidin-7-yl)aminokvcl nent\l)methyl sulfamate
[0165] To a solution of ((3aR,4R,6R!6aS)-2,2-dimethyl-6-((2-(3- ((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (2.0 g) in acetonitrile (11 mL) at 0°C was added phosphoric acid (1 1 mL) while maintaining the temperature below 10°C. This mixture was warmed to ambient temperature and stirred for 4 hours. At this time HPLC analysis showed that <1% ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate starting material or reaction intermediates remained. To the reaction was added ethyl acetate (1 1 mL) and water (11 mL) and saturated Na2C03 (10 mL) dropwise. After this addition was complete saturated Na2C03 was added until the pH was between 6-7. The phases were separated and to the organic layer was added acetonitrile (30 mL) and the mixture was concentrated on a rotovap to ~16 mL. The mixture was stirred overnight. During this time a white slurry formed. The white solids were filtered using a medium filtted filter. The solid was dried in a vacuum oven at full vacuum overnight (40°C). The reaction was then analyzed by HPLC and NMR to give ((lR,2R,3S,4R)-2,3-dihydroxy-4-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)cyclopentyl)methyl sulfamate (1.5g ,84%). lH NMR (300 MHz, DMSO-c¾) δ ppm 1.44 - 1.61 (m, 1 H) 2.20 - 2.42 (m, 2 H) 3.78 (q, J-4.50 Hz, 1 H) 3.90 - 4.09 (m, 3 H) 4.09 - 4.22 (m, 1 H) 4.80 (d, ^5.28 Hz, 1 H) 5.03 (d, J=5.28 Hz, 1 H) 6.31 (d, J=5.57 Hz, 1 H) 7.05 (s, 1 H) 7.48 (s, 2 H) 7.62 - 7.72 (m, 1 H) 7.77 (d, J=7.92 Hz, 2 H) 8.17 (d, J=5.28 Hz, 1 H) 8.31 (dt, ^7.70, 1.43 Hz, 1 H) 8.47 (s, 1 H).
[0166] Example 3: fflR.2R.3S.4RV2.3-dihvdrosy-4-ff2-f3- ( ( trifluoroniethyl )thio)ph en vDpyrazolo 11,5-a I pyi Lmidin-7-Yl)amino)cvclopcntyl>m ethyl sulfama te
[0167] Step 1: .2-dimethyl-5-ff -(3-frtrifluoromethvnthio)phenvn-lH-pyrazol-5-yl)ainino)methylene -l,3-dioxane-4,6-dione)
[0168] Under a blanket of nitrogen at 20°C, Meldrum's acid (18.6 Kg) and isopropanol (33 L) were placed in a 100 L glass-lined reactor. Trimethyl orthoformate (15.5 Kg (16.0L)) and isopropanol (11 L) were added and the mixture was heated to 80 °C for 40 min, whereby a small amount of methanol distilled off (<0.5 L). The mixture was stirred for 2 h at 80 °C. in a separate 160 L glass-lined reactor under nitrogen at 20 °C, 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (prepared in the manner described above) was mixed with isopropanol ( 10.9 kg, 42.0 mmol) and heated up to 80 °C within 60 min. The content of the 100 L reactor was transferred into the reaction mixture in the 160 L reactor at 80 °C, which was completed after 3 min. The reaction mixture was stirred for 30 min at 78 °C, the reaction was then cooled to 60 °C. HPLC analysis showed the reaction was 99.56% complete (product%/(product%+starting material0/.). The reaction mixture was cooled to 20 °C within 100 min, then the mixture was stirred for further 100 min at 20 °C. The suspension was then transferred onto a pressure filter. At 1.2 bar nitrogen, the solids were collected on the filter. The filter cake was washed 4 x with ethyl acetate (18 L each time). The wet cake was dried on the filter for 17 h at 20°C using a slight stream of nitrogen/vacuum (200-100 mbar). The wet product (14.7 kg) was further dried at the rotavap for approx. 24 h at 40-50 °C. 11,75 kg of the crude title compound was obtained (68% yield). NMRspectrum was consistent with that described above in Example 2.
[0169] Step 2: 2-(3-fftrifluoromethvnthio)phenYnpyrazolori.S-a1pyrimidin-7-ol
[0170] Under nitrogen at 20 °C, (2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l ,3-dioxane-4,6-dione) was placed in the reactor. 1 ,2-Dichlorobenzene (117 L) was added. The suspension was heated to 147°C for 90 min to give a solution, then it was stirred at 147°C for 18 h. Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 92.28% completion (product%/(product%+starting material%). The mixture was heated up again to 147°C and stirred for further 5 h at this temperature. HPLC analysis showed the reaction was 96.51% complete (product%/(product%+starting material%). The mixture was then stirred for 48 hours at 20°C, then it was heated again to 147°C und stirred at this temperature for 5 h. Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 98.47% completion (product%/(product%+starting material%). The mixture was heated up again to 146°C and stirred for further 5 h at this temperature.
Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 99.35% complete (product%/(product%+starting material%). The reaction was cooled to 20°C and the suspension was transferred in a pressure filter. The solids were collected on the filter at 1.8-3 bar N2 over a greater than 10 hour period. The filter cake was washed 4 x with acetonitrile (17 L), then it was dried on the filter for 18 h at 20°C/200-100 mbar, using a slight stream of N2. The material was transferred to a 50 L flask and dried on a rotavap at 50-60°C / 24-14 mbar for 2 d. 6.118 kg of the crude title compound was obtained (70% yield). NMR spectrum was consistent with that described above in Example 2.
[0171] Step 3: l-f2-f3- trifluoromethYnthio^phenvnpyrazoIo[1.5-alpyriinidiii-7-vn-lH-benzofdl [1.2.31 triazolc: triethylamine: hydrochloride complex ( 1 :0.21:0.21 moles:moles:moles)
[0172] Under N2 at 20°C, acetonitrile (30 L) was placed in the reactor, 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (6.00 kg) and lH-benzotriazol (5.83 kg) was added. A further portion of acetonitrile (30 L) was added, then the mixture was stirred at 20°C. Stirring proceeded over night. Triethylamine (8.16 L) was added at 20°C over 6 min. The yellow suspension was heated up to 45°C for 40 min. While stirring at 150 rpm, phosphoryl chloride (4.562 kg) was slowly added for 45 min. By controlling the addition, the reagent was dropped directly into the mixture to avoid the formation of lumps. The addition was exothermic, a maximum temperature of 53°C was observed. The brown suspension was heated up to 80°C over 1 h, then the reaction mixture was stirred for 5 h at this temperature. Acetonitrile (30 L) was added over 20 min keeping the internal temperature between 75-80°C. HPLC analysis showed the reaction was 98.31% completion (product%/(product%+starting material%).The mixture (brown suspension) was further stirred at 80°C for 70 min. HPLC analysis showed the reaction was 99.48% completion (product%/(product%+starting material%). Acetonitrile (61 L) was added over 30 min maintaining the temperature between 75-80°C. The pale brown suspension was stirred at 80°C for 90 min, then it was cooled to 20°C over 2.5 h. The mixture was stirred for 12 h at 20°C. The mixture was transferred in a pressure filter. The filter cake was washed twice with acetonitrile ( 18 L). Both wash steps were done at 3.5-4 bar N2. Each of these filtrations took overnight to go to completion. The filter cake was dried on the filter for 7.5 h. The material was transferred in a 50 L flask and dried at the rotavap at Ta 40-50°C / 50-11 mbar for 3 d to get a dry mass of 99.88% . The yield of l -(2-(3-((trifluoromethyl)t]iio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-yl)-lH-benzo[d][l,2,3]triazole: triethylamine: hydrochloride complex (1 :0.21 :0.21 moles:moles:moles) was 7.948 kg (75%). NMR spectrum was consistent with that described above in Example 2.
[0173] Step 4: 3aR4R.6R,6aS)-2,2-dimethYl-6-f(2-f3-ffMfluoromethvnthio phenvnpyrazolori.5-alDyrimidin-7-yl)amino)tetrahvdro-3aH- vclopenta Idl [1.31 dioxol-4-vDmethanol
[0174] Under N2 in a 160 L glasslined reactor, triethylamine (21%) compound with l -(2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5 -a] pyrimidin-7-yI) - 1 H-benzo [d] [ 1 ,2,3 Jtriazole (21 %) hydrochloride (7.86 kg) was dissolved in triethylamine (23.3 L) at 20°C. ((3aR,4R,6R,6aS)-6-amino-2,2-dimethyltetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol hydrochloride (4.49 kg) was added, followed by triethylamine (23 L). The reaction mixture was heated up to 80°C over 1 h, and then the mixture was stirred for 8 h at 80°C. The mixture was then cooled to 20°C. HPLC analysis showed the reaction was 99.97% complete (product%/(product%+starting material%). Water (66 L) was then added over 30 min at 20-25°C (exotherm), whereby a brown suspension was obtained. The mixture was concentrated at 60°C, 150-95 mbar, until 42 L solvent was distilled off. The suspension was heated to 50°C, and the solids were collected on a 90 L pressure filter (1.2 bar N2), which took 40 min. During this process, the material on the filter was not actively heated. The remaining solids in the reactor were rinsed with 15 L of the mother liquor. The wet filter cake was transferred back in the reactor. Water (64 L) was added. The mixture was heated up to 50°C over 30 min. The washed solids were collected on the 90 L pressure filter. Remaining mother liquor in the filter cake was pressed off at 1.2 bar N2 for 50 min (50 L mother liquor was used to rinse the reactor). The filter cake was dried on the pressure filter for 13.5 h, applying a slight stream of N2 / vac at 20°C to afford 10.247 kg of crude ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)tWo)phenyl)pyrazolo[l ,5-a]pyrimidin-7-yl)ammo)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol. The wet filter cake was isolated. The wet filter cake was loaded into the reactor. Acetonitrile (65 L) was added, followed by activated charcoal (6.59 kg). The mixture was heated to 50°C for 30 min and stirred for 2 h at 50°C. Meanwhile a bed of celite (4.25 kg) had been prepared in the 90 L pressure filter, using acetonitrile (20 L) for conditioning. The bed was heated at 50°C. The black suspension was transferred on the filter and pushed through the Celite plug at 2 bar. The filtrate was transferred to a 200 L stirring tank via a heat resistant tube and a 0.45 μιη inline filter. The operation needed 18 min for completion. For washing, acetonitrile (50 L) which had been warmed up in the reactor to 50°C and transferred over the warmed filter cake and pushed through at 2 bar. Again, the filtrate was transferred in the 200 L stirring tank via a heat resistant tube and a 0.45 μιη inline filter. The operation needed 10 min for completion. The reactor was cleaned to remove attached charcoal (abrasive cleaning, using NaCl /acetone). The filtrate in the stirring tank was transferred in the reactor and concentrated at 50°C / 120 mbar until 63 L were distilled off. While well stirring (300 rpm) and 50°C, Water (1 10 L) was slowly added over 2 h. A pale yellow suspension was formed. The concentrate was cooled to 20°C for 3 h, then stirred at this temperature for 13 h. The solids were collected on a 50 L filter, using 1.2 bar N2 to push the filtrate through. The filter cake was washed twice with water (18 L), then dried on the filter for 24 h at 200-100 mbar, using a slight stream of N2. 4.563 kg of the title compound was obtained 55% yield. NMR spectrum was consistent with that described above in Example 2.
[0175] Step 5: (f3aR,4R,6R,6aS)-2^-dimethyl-6-(f2-f3-fftrifluorQmethvnthio phenvnpyrazolo[1.5- |pyrimidm-7-vnamino)teti ahYclro-3aH-cvclopenta|d||1.3ldioxol-4-yl mcthyl tert-butoxycarbonylsutfamatc
[0176] Under N2 at 20°C, ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3- ((trifluoromethyl)thio)phenyl)pyrazolo[ 1 , 5 -a]pyrimidin-7-yl)amino)tetrahydro-3 aH-cyclopenta[d][l,3]dioxol-4-yl)methanol (4.019 kg) was placed in a 160 L glasslined reactor, then 2-methyl-tetrahydrofuran (40 L) was added. The mixture was stirred at 150 rpm for 30 min at 20°C, whereby a clear solution was formed. A KF measurement was taken and showed the water content to be 0.036% H20. The solution was stirred over night at 20 °C. The next morning, PPTS (2.2 kg) was loaded into the reactor. At 20°C, (4-aza-l-azoniabicyclo[2.2.2]oct-l-yIsulfonyl)(tert-butoxyc£u-bonyl)azanide-l,4-diazabicyclo[2.2.2]octane (1:1) hydrochloride (10.2 kg) was added. Stirring of the heterogeneous mixture was started at 130 rpm. The reaction was stirred with 200 rpm for 1 h at 20°C, then with increased speed of 250 rpm for an additional hour. HPLC analysis showed the conversion to be 87.3%. The reaction mass was stirred with 300 rpm for 2 h at 20°C. HPLC analysis showed the conversion to be 95.6%. The reaction mass was stirred with 300 rpm for 2 h at 20°C. HPLC analysis showed the conversion to be 97.7%. NaHC03 3.7% (40 L) was added to the mixture at 20°C and the reaction was stirred at 300 rpm for 10 min. Most of the solids from the reaction mixture went into solution. To dissolve remaining material which was attached at the top of the reactor, the bilayered mixture was stir up shortly by a N2 stream from the bottom. The layers were separated, which was completed after 13 min. The aqueous layer was discharged, the organic layer remained in the reactor. The org. layer was a brown solution, the aqueous layer was colorless and turbid. The pH of aqueous layer was approx. 8 (pH stick). NaHC03 3.7% (40 L) was added to the mixture at 20°C and it was stirred at 300 rpm for 10 min. The layers were separated, which was completed after 27 min. The aqueous layer was discharged, the organic layer remained in the reactor. The organic layer was a brown solution, the aqueous layer was colorless and turbid. The pH of aqueous layer was approx. 8-9 (pH stick) and the pH of organic layer was approx. 8 (pH stick, wet). The product in organic layer was transferred in the feeding tank and stored temporarily (approx. 30 min) at 20°C. The reactor was optically cleaned using a mixture of 2-methyltetrahydrofuran (30 L) and H20 (20 L). The org. layer was placed in the reactor and stored at -20°C for 14.5 h . While stirring at 150 rpm, the org. layer (suspension) was diluted with acetonitrile (16 L) and water (15 L) and warmed up to 5°C. At 5°C, acetic acid (0.172 kg) was added over 5 min. to a pH of 6; resulting in a mixture that was a pale brown solution. ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (2.0 g; prepared in a similar manner to that described above Example 2, Step 5) was added as seed. At 5°C, acetic acid (0.515 kg) was added over 15 min. to pH 4-5; a suspension formed. The feeding tank was rinsed with water (1.6 L). The mixture was stirred at 5°C with 90 rpm for 1.5 h, then it was transferred in a 50 L filter and filtered at 1.2 bar N2, in only 4 min. The filter cake was washed 4 x with cold acetonitrile (8 L, 0-5°C), then it was dried on the filter at 20°C for 8 h at 200 mbar, using a slight stream of N2. The yield of the title compound was 3.594 kg (62%). MR spectrum was consistent with that described above in Example 2.
[0177] Step 6: friR.2R.3S.4R 2.3-dihvdroxY-4-ff2-f3-fftrifluoromethvntliio phenvnDyrazolori.5-alpyrimidin-7-yl)aminokvciopent>T)mcthyl sulfamate Compound 1
[0178] 3.538 kg of ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate was suspended in 13.5 kg of acetonitrile and cooled to 5°C. To this mixture was added 27.3 kg of H3PO4 over 1 hour and 50 minutes. The reaction was warmed to 20°C over 50 minutes and then stirred for 8h at 22°C. HPLC analysis showed the reaction was 99.69% complete. To the first portion (50% of the reaction mixture) was added 8.9 kg of water and 7.95 kg of ethyl acetate. The pH was then adjusted to 6.5 with 48 L of saturated sodium carbonate. 7.7 kg of ethyl acetate was added and the phases were separated. To the second portion (50% of the reaction mixture) was added 8.9 kg of water and 7.95 kg of ethyl acetate. The pH was then adjusted to 6.15 with 48 L of saturated sodium carbonate. 7.7 kg of ethyl acetate was added and the phases were separated. The organic phases were combined in a vessel (rinsed with 1.8 kg of ethyl acetate) and washed with 17.8 kg of water. The phases were separated and 17.8 kg of water and 0.237 kg of NaCl were added and the phases were separated. A repeat of wash with 17.8 kg of water and 0.237 kg of NaCl was added and the phases were separated. The organic layers were then combined and the temperature of the mixture was raised to 40°C and the pressure was reduced to 300-142 mbar. 27 L of liquid was distilled off over 4h. 31.7 kg of acetonitrile were then added to the solution and the temperature of the mixture was raised to 38°C and the pressure was reduced to 320-153 mbar. 26 L of liquid was distilled over 3h. 31.7 kg of acetonitrile were then added to the solution and the temperature of the mixture was raised to 37°C and the pressure was reduced to 320-153 mbar. 34 L of liquid was distilled over 2h. The suspension was stirred for lh at 50°C and then cooled to 20-25°C over 3h. The reaction was stirred overnight and the product was filtered and washed with 8.9 kg of acetonitrile twice. The cake was dried for 2h at 20°C (33 mbar) then at 40-45°C (1 mbar) to afford 2.08 kg (75.8%) of the title compound. 2.066 kg of ((lR,2R,3S,4R)-2,3-dihydroxy-4-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 , 5 -a]pyrimidin-7-yl)amino)cyclopenty l)methy 1 sulfamate was loaded into a reactor with 9.76 kg of acetronitrile and 4.12 kg of water and heated at a temperature of 56 °C for 1 hour and 10 minutes until dissolved. The solution was polished filtered and the filter was
rinsed with 3.16 kg acetonitrile and 1.37 kg of water. To the resulting solution was added with 11.0 kg of water over 45 minutes while maintaining the reaction temperature between 52-55°C. 0.009 kg of (( 1 R,2R,3S,4R)-2,3 -dihydroxy-4-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)cyclopentyl)methyl sulfamate was added as seed (prepared in a similar manner to that described above Example 2, Step 5). A suspension was visible after 10 minutes of stirring. To the solution was added 9.62 kg of water over 3h while maintaining the reaction temperature between 50-55°C. The suspension was then cooled over 3h to 20°C and stirred for 12h at 22-23°C. The suspension was then filtered and washed twice with 13.7 kg of water. The product was dried at 40°C. 1.605 kg of the title compound was obtained in 78% yield. NMR spectrum was consistent with that described above in Example 2.
PATENT
WO2016069392
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016069392&recNum=162&docAn=US2015057062&queryString=FP:(%22cancer%22)%20AND%20EN_ALL:nmr&maxRec=28697
SYNTHESIS

///////////////1450833-55-2,
MLN 7243, TAK-243, TAK 243, TAK243, MLN7243; MLN-7243, MLN 7243,
AOB87172, AOB-87172, AOB 87172, Millennium Pharmaceuticals, Inc., PHASE 1, TAKEDA ONCOLOGY
COS(=O)(=O)N[C@H]1C[C@H]([C@@H]([C@@H]1O)O)NC2=CC=NC3=CC(=NN23)C4=CC(=CC=C4)SC(F)(F)F