
Liarozole
CAS Registry Number: 115575-11-6
CAS Name: 5-[(3-Chlorophenyl)-1H-imidazol-1-ylmethyl]-1H-benzimidazole
Additional Names: (±)-5-(m-chloro-a-imidazol-1-ylbenzyl)benzimidazole
Molecular Formula: C17H13ClN4
Molecular Weight: 308.76
Percent Composition: C 66.13%, H 4.24%, Cl 11.48%, N 18.15%
Melting point: mp 108.2°
Derivative Type: Fumarate
CAS Registry Number: 145858-52-2
Manufacturers' Codes: R-85246
Trademarks: Liazal (Janssen)
Molecular Formula: 2C17H13ClN4.3C4H4O4
Molecular Weight: 965.75
Percent Composition: C 57.21%, H 3.97%, Cl 7.34%, N 11.60%, O 19.88%
Derivative Type: Hydrochloride
CAS Registry Number: 145858-50-0
Manufacturers' Codes: R-75251
Molecular Formula: C17H13ClN4.HCl
Molecular Weight: 345.23
Percent Composition: C 59.14%, H 4.09%, Cl 20.54%, N 16.23%
Therap-Cat: Antineoplastic.


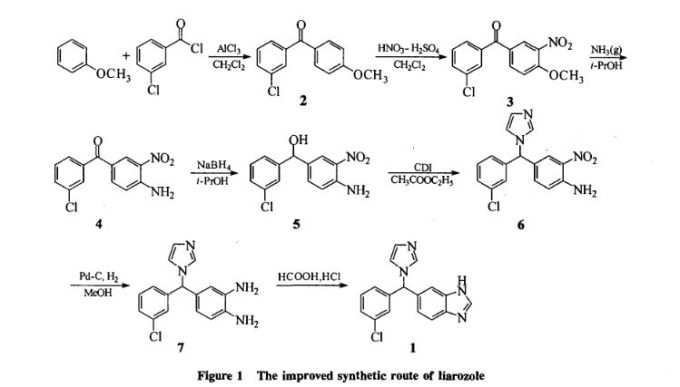
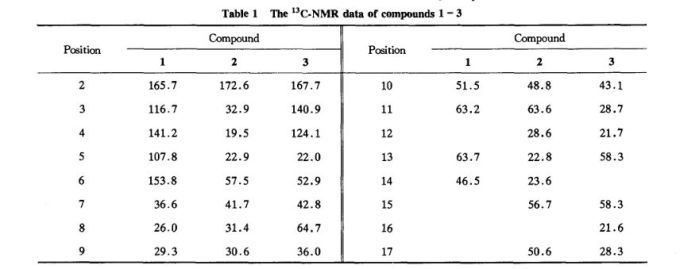
Liarozole fumarate is prepared as shown in Scheme 20970301a. Anisol is reacted with 3-chlorobenzoyl chloride (I) under Friedel-Craft conditions to give (3-chlorophenyl)(4-methoxyphenyl)methanone (II). Nitration of (II) is carried out in dichloromethane at 10 C to yield (III). The methoxy group in (III) is replaced by the amino group by means of NH3 in 2-propanol at 100 C under pressure, giving (IV). By reduction of the keto function of (IV) with sodium borohydride in 2-propanol, the corresponding alcohol (V) is obtained, which upon treatment with 1,1'-carbonyldiimidazole in refluxing dichloromethane yields the imidazolyl compound (VI). Hydrogenation of the nitro group in (VI), followed by cyclization of (VII) in a refluxing mixture of formic acid and 4N hydrochloric acid, gives the benzimidazole derivative (VIII). Finally, the treatment of (VIII) with fumaric acid in ethanol yields liarozole fumarate (IX).
http://www.google.com/patents/WO1995022540A1?cl=en
Liarozole is a racemic mixture, i.e. a mixture of its optical isomers, and is specifically mentioned as compound 28 in EP-0,371,559. Said patent application mentions the use of compounds like liarozole in the treatment of epithelial disorders. EP-0,260,744 describes the use of compounds like liarozole for inhibiting or lowering androgen formation. Whereas EP-0,371,559 and EP-0,260,744 recognize that compounds like liarozole have stereochemically isomeric forms, no example of an enantiomerically pure form is given of liarozole.
Chemically liarozole is (±)-5-[3-chlorophenyl]-lH-imidazol-l-ylmethyl]-lH-benz- imidazole, and is represented by formula (I). As can be seen from the chemical structure, liarozole has one stereogenic center (indicated with an asterisk in formula (I)).

General preparation of structures including liarozole have been extensively described in EP-0,371,559 and EP-0,260,744.
Enantiomerically pure (+)-liarozole may be prepared by reacting an enantiomerically pure intermediate diamine of formula (B)-(II) with formic acid or a functional derivative thereof.

The general reaction conditions, work-up procedures and conventional isolation techniques for carrying out the above and following reactions are described in the prior art. When more specific conditions are required they are mentioned hereinunder. The enantiomerically pure intermediate diamine of formula (B)-(II) may be prepared by reducing an intermediate of formula (B)-(iπ) by a standard nitro-to-amine reduction reaction.

Appropriate solvents for carrying out said fractional crystallization are water, ketones, e.g. 2-propane, 2-butanone; alcohols, e.g. methanol, ethanol, 2-propanol. Mixtures of ketones and water are very suitable for the above fractional crystallization. Preferably a mixture of 2-propanone and water is used.
The ratio of water/2-propanone by volume may vary from 1/10 to 1/2. Preferred range of said ratio is 1/5 to 1/3.
The fractional crystallizations are suitably carried out below room temperature, preferably below 5°C.
It was also found that the subsequent reaction step can be carried out without any appreciable racemization.
Alternatively the (+)-isomer of the compound of formula (I) may be prepared by cyclizing an intermediate of formula (B)-(IV) following procedures as described above for the cyclization of intermediates of formula (B)-(II) and desulfurating the thus obtained intermediate of formula (B)-(V). In formulas (B)-(TV) and (B)-(V) R represents Ci^alkyl, wherein Ci-^alkyl means a straight or branch chained saturated hydrocarbon radicals having 1 to 6 carbon atoms such as, for example, methyl, ethyl, propyl, butyl, pentyl, hexyl. Preferably R is methyl.

(B)-(D ), and reducing the nitro group of the intermediate (B)-(X). In the formulas
(Vπ), (B)-(Vm), (B)-(IX) and (B)-(X) R represents Ci^alkyl as defined hereinabove.
S OR

Experimental part
A. Preparation of the intermediates
Example 1 a) A heterogeneous mixture of (±)-4-[(3-chlorophenyl)-lH-imidazol-l-ylmethyl]-2- nitrobenzenamine (the preparation of which is described in EP-371,559) (500 g) in
2-propanone (2000 ml) and water (100 ml) was stirred at 22°C. (-)-(lR)-7,7-dimethyl- 2-oxo-bicyclo[2.2.1]heptane-l-methanesulfonic acid (353.2 g) was added and the mixture became homogeneous after 10 minutes. The mixture was first stirred for 18 hours at 20°C and then for 3 hours at 0-5°C. The precipitate was filtered off, washed with 2-propanone/water 95/5 (150 ml) and dried, yielding 308.9 g (36.2%) of product A sample (306.7 g) was partitioned between dichloromethane (500 ml) and water (750 ml). Ammonium hydroxide (100 ml) was added. This mixture was stirred for 15 minutes. The aqueous layer was separated and extracted twice with dichloromethane (250 ml each time). The separated organic layer was washed with water (250 ml), dried, filtered and the solvent was evaporated, yielding 179.7 g of (-)-(B)-4-[(3-chlorophenyl)-
20 lH-imidazol-l-ylmethyl]-2-nitrobenzenamine; mp. 89.8°C; [α]D = -19.80° (c = 0.5% in methanol) (interm. 1). b) A mixture of intermediate (1)(179.7 g) in methanol (656 ml) and a solution of ammonia in methanol (32.7 ml) was hydrogenated at 20-25 °C with platinum on activated carbon (13.1 g) as a catalyst in the presence of thiophene (0.27 g). After uptake of hydrogen (3 eq.) the catalyst was filtered off and washed with 2-propanol (30 ml). A solution of hydrochloric acid in 2-propanol (522 ml) was added to the filtrate at <30°C. The mixture was stirred for 3 hours at 20 °C, then for 3 hours at 0-5 °C. The resulting precipitate was slowly filtered off, washed with methanol (100 ml) and dried
(50 °C), yielding 185.60 g (83.2%) (+)-(B)-4-[(3-chlorophenyl)-lH-imidazol-l-yl-
20 methyl]- 1,2-benzenediamine trihydrochloride; mp. 172.5°C; [α^ = +23.73° (c = 1% in methanol) (interm. 2).
Example 2 a) A mixture of (4-amino-3-nitrophenyl) (3-chlorophenyl)methanone (50 g), formamide (375 ml) and formic acid (63 ml) was stiιτed and refluxed for 17 hours. After cooling, the mixture was poured on ice. The precipitate was filtered off and dried, yielding 55 g (99.4%) of (±)-N-[(4-amino-3-nitrophenyl) (3-chlorophenyl)methyl]formamide (interm. 3). b) A mixture of intermediate (3) (50.7 g), hydrochloric acid 6N (350 ml) and 2-propanol (70 ml) was stirred and refluxed for 17 hours. The yellow precipitate was filtered off and dried in vacuo, yielding 51 g (97.8%) of (±)-4-amino-α-(3-chloro- phenyl)-3-nitrobenzenemethanamine monohydrochloride; mp. 263°C (interm.4). c) To a solution of intermediate (4) (43 g) in tetrahydrofuran (400 ml) at room temperature was added succesively N,N-diethylethanamine (13.8 g) and (R)-(-)-α- hydroxybenzeneacetic acid (20.8 g). Then a solution of 1-hydroxybenzotriazole monohydrate (22.2 g) in tetrahydrofuran (200 ml) was added. After complete addition a solution of N,N'-dicyclohexylcarbodiimide (33.9 g) in dichloromethane (300 ml) was introduced to the mixture. After stirring for 2 hours at room temperature N,N'- dicyclohexylurea was filtered off. The filtrate was washed with a solution of potassium carbonate (10%) and the organic layer was dried to give a mixture of diastereomers (60g) (fraction 1). The same experiment with intermediate (4) (16 g) as starting material resulted in a yield of 26 g of a mixture of diastereomers (fraction 2). Fraction 1 and 2 were combined and purified by HPLC (eluent : CH2θ2/ethyl acetate 90:10), yielding 30g (32.3%) of (±)-(R,B)-N-[(4-amino-3-nitrophenyl)(3-chlorophenyl)methyl]-α- hydroxybenzeneacetamide (interm.5). d) A mixture of intermediate (5) (30 g), hydrochloric acid 12N (300 ml) and 1-propanol (100 ml) was stirred and refluxed for 17 hours and poured on ice. The mixture was extracted with ethyl acetate. The aqueous phase was basified with ammonium hydroxide and extracted with dichloromethane. The dichloromethane extracts were dried, filtered and evaporated, yielding 7.3 g (36.0%) of (+)-(B)-4-amino-α-(3-chlorophenyl)-3- nitrobenzenemethanamine (interm. 6). e) A mixture of intermediate (6) (7.3 g), 2-isothiocyanato-l,l-dimethoxyethane (4.8 g) and methanol (75 ml) was stirred and refluxed for 2 hours. The mixture was evaporated to an oily residue, yielding 11 g (100%) of (+)-(B)-N-[(4-amino-3-nitrophenyl)(3- chlorophenyl)methyl]-N'-(2,2-dimethoxyethyl)thiourea (interm.7). f) A mixture of intermediate (7) (11 g), iodomethane (2 ml) and potassium carbonate (4.97 g) was stirred at room temperature for 48 hours. The solvent was evaporated and the residue was taken off with dichloromethane and washed with water. The organic layer was dried, filtered and evaporated, yielding 11.4 g of (+)-(S)-methyl (B)-N- [(4-amino-3-nitrophenyl)(3-chlorophenyl)methyl]-N'-(2,2-dimethoxyethyl)carbam- imidothioate as an oily residue (interm. 8). g) To intermediate (8) (11.4 g) at 0°C was added sulfuric acid (100ml) (precooled to 5°C). The mixture was stirred at 5°C until complete dissolution and then was warmed to room temperature. After stirring for 2 hours, the solution was poured on ice and basified with ammonium hydroxide. The aqueous solution was extracted with ethyl acetate. The organic layer was dried, filtered and evaporated. The residue was purified by column chromatography (eluent : CH2CI2/CH3OH 98:2). The eluent of the desired fraction was evaporated, yielding 3.7 g (38.0%) of (+)-(B)-4-[(3-chlorophenyl)[2-(methylthio)-lH- imidazol-l-yl]methyl]-2-nitrobenzenamine (interm.9). h) A mixture of intermediate (9) (6.2 g), Raney nickel (6 g) and methanol (100 ml) was hydrogenated for 2 hours at 2 bar and at room temperature. After the calculated amount of hydrogen was taken up, the catalyst was filtered off. The filtrate, (+)-(B)-4-[(3- chlorophenyl)[2-(methylthio)-lH-imidazol-l-yl]methyl]-l,2-benzenediamine (interm. 10), was used for the next step. i) A mixture of intermediate (10) (5.7 g), methanimidamide monoacetate (5.2 g) and methanol (100 ml) was stirred and refluxed for 3 hours. The reaction mixture was evaporated and the residue was taken off in dichloromethane and washed with sodium hydrogen carbonate (10%). The organic layer was dried, filtered and evaporated. The oily residue was purified by column chromatography (eluent : CH2CI2/CH3OH 95:5). The eluent of the desired fraction was evaporated, yielding 4.9 g (83.7%) of (+)-(B)-5-[(3-cWorophenyl)[2-(methylthio)-lH-imidazol-l-yl]methyl]-lH-benzimidazole (interm. 11).
B. Preparation of the final compounds Example 3
A mixture of intermediate (2) (185 g) in water (512 ml) was stirred at 20 °C. Hydrochloric acid (289 ml) was added. Formic acid (85%) (61.17 ml) was added and this mixture was heated to 55°C. The reaction mixture was stirred for 3 hours at 55 °C and then cooled to 20°C. Dichloromethane (1223 ml) was added. Ammonium hydroxide (730 ml) was added dropwise at < 25°C. The separated organic layer was washed with water (500 ml), dried, filtered and the solvent was evaporated, yielding 152.88 g (108.5%) of product. A sample was dried (18 hours at 55 °C), yielding 3.18 g of (+)-(B)-5-[(3-chlorophenyl)-lH-imidazol-l-ylmethyl]-lH-benzimidazole; mp.
20 113.7°C; [αjj = +43.46° (c = 1% in methanol) (comp. 1).
Example 4
A mixture of intermediate (11) (4.9 g), Raney nickel (2 g) and ethanol (100ml) was stirred and refluxed for 5 days, while every day an additional amount of Raney nickel (2 g) was added. The catalyst was filtered off and rinsed with dichloromethane. The filtrate was evaporated and the residue was purified twice by column chromatography (silica gel; CH2CI2/CH3OH 95:5 ; CH2CI2/CH3OH NH4OH 80:20:3). The eluent of the desired fraction was evaporated and the residue was converted into the hydrochloride salt in 2-propanol and ethanol. The salt was recrystallized from 2-butanone, yielding 1.8 g (37.2%) of (+)-(B)-5-[(3-chlorophenyl)(lH-imidazol-l-yl)methyl]-lH-benzimidazole
20 monohydrochloride; mp. 212.1°C; [α]D = +42.43° (c = 1% in ethanol) (comp. 2)
Example 5
Compound (1) (149.7 g) was dissolved in 2-butanone (2424 ml). A mixture of hydrochloric acid in 2-propanol (82.6 ml) in 2-butanone (727 ml) was added over a 2 hour period at 20 °C. The reaction mixture was stirred for 16 hours at 20 °C. The precipitate was filtered off, washed with 2-butanone (242 ml) and dried (vacuum; 80°C); yielding 147.5 g (99.3%) of (+)-(B)-5-[(3-chlorophenyl)-lH-imidazol-l-ylmethyl]-lH-
20 benzimidazole monohydrochloride; mp. 214.5°C; [α] j = +36.20° (c = 1% in methanol) (comp. 2). Example 6
A mixture of compound (1) (0.72 g) in ethanol (5.1 ml; denaturated) was stirred at 20 °C until it became homogeneous. (E)-2-butenedioic acid (0.54 g) was added The mixture was stirred for 18 hours at 20 °C and then cooled 0-5 °C and precipitation resulted. More denaturated ethanol (2 ml) was added and the mixture was stirred for 2 hours at 20 °C. The precipitate was filtered off, washed with ethanol (3 ml; denaturated) and dried (vacuum; 50 °C), yielding 0.26 g (23.4%) (B)-5-[(3-chlorophenyl)-lH-imidazol-l-yl- methyl]-lH-benzimidazole (E)-2-butenedioate (2:3).ethanolate (2:1); mp. 111.2°C (comp. 3).
PAPER
Improved synthesis of liarozole
J Ren, Y Sha, D Zhao, M CHENG - Chinese Journal of Medicinal …, 2006 - en.cnki.com.cn
... 1-yl)-methyl]-1H-benzimidazole(liarozole).Methods Starting from anisole,liarozole was synthesized
by Friedel-Crafts(acylation,)nitration,nucleophilic substitution,reduction and cyclization.Results
and conclusion The structure of liarozole was confirmed by()~1H-NMR and MS ...
see at..http://lib.syphu.edu.cn/71%E6%A0%A1%E5%86%85%E7%BD%91%E4%B8%93%E7%94%A8/zwlw%E5%85%A8%E6%96%87/60230.pdfby Friedel-Crafts(acylation,)nitration,nucleophilic substitution,reduction and cyclization.Results
and conclusion The structure of liarozole was confirmed by()~1H-NMR and MS ...
Paper
Conversion of the Laboratory Synthetic Route of the N-Aryl-2-benzothiazolamine R116010 to a Manufacturing Method
Chemical Process Research Department, Janssen Pharmaceutica, Turnhoutseweg 30, 2340 Beerse, Belgium
Org. Proc. Res. Dev., 2001, 5 (5), pp 467–471
DOI: 10.1021/op0100201
PAPER
Synthesis and In Vitro Evaluation of3-(1-Azolylmethy1)-1H-indolesand
341-Azolyl-l-phenylmethyl)-1H-indolesasInhibitorsofP450arom
https://www.researchgate.net/profile/Marc_Le_Borgne/publication/8068537_2-_and_3-%28aryl%29%28azolyl%29methylindoles_as_potential_non-steroidal_aromatase_inhibitors/links/02e7e52fe95f662b24000000.pdf
Literature References: Inhibits cytochrome P450-dependent enzymes involved in steroid biosynthesis and retinoic acid catabolism. Prepn: A. H. M. Raeymaekers et al., EP 260744; eidem, US 4859684 (1988, 1989 both to Janssen). In vivo antitumor activity: R. Van Ginckel et al., Prostate 16, 313 (1990). Pharmacology and effect on steroid synthesis: J. Bruynseels et al., ibid., 345; and effect on retinoic acid: R. De Coster et al., J. Steroid Biochem. Mol. Biol. 43, 197 (1992). Clinical evaluation in prostate cancer: C. Mahler et al., Cancer 71, 1068 (1993); in psoriasis: P. Dockx et al., Br. J. Dermatol. 133, 426 (1995); in combination therapy for malignant brain tumors: M. E. Westarp et al., Onkologie 16, 22 (1993).
 | |
| Names | |
|---|---|
| IUPAC name
6-[(3-Chlorophenyl)-imidazol-1-ylmethyl]-1H-benzimidazole
| |
| Identifiers | |
| 115575-11-6 | |
| ChemSpider | 54664 |
| 5210 | |
| Jmol interactive 3D | Image |
| PubChem | 60652 |
| Properties | |
| C17H13ClN4 | |
| Molar mass | 308.77 g·mol−1 |
C1=CC(=CC(=C1)Cl)C(C2=CC3=C(C=C2)N=CN3)N4C=CN=C4
No comments:
Post a Comment