CONAZOLE

1 TERCONAZOLE
2 KETOKONAZOLE 2S 4R
3 ISAVUCONAZOLE
4 TIOCONAZOLE
5 FOSRAVUCONAZOLE
6 RAVUCONAZOLE
7 EFINACONAZOLE
8 ALBACONAZOLE
9 BUTOCONAZOLE
PART 1..........http://drugsynthesisint.blogspot.in/p/conazole.html
PART 2
10 VORICONAZOLE
11 POSACONAZOLE
12
PART 2

10
VORICONAZOLE
 .
.
VORICONAZOLE
CAS 137234-62-9
(2R,3S)-2-(2,4-difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1,2,4-triazol-1-yl)butan-2-ol
Voriconazole (vor-i-KON-a-zole, brand name Vfend, Pfizer) is a triazole antifungal medication that is generally used to treat serious, invasive fungal infections. These are generally seen in patients who are immunocompromised, and include invasive candidiasis, invasive aspergillosis, and certain emerging fungal infections



1H NMR......... http://file.selleckchem.com/downloads/nmr/S144202-Voriconazole-HNMR-Selleck.pdf
1H NMR DMSO-d6, peak at 3.3 is HOD

13 C NMR
DMSO-d6

CAS 137234-62-9
(2R,3S)-2-(2,4-difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1,2,4-triazol-1-yl)butan-2-ol
| Chemical Names: | Voriconazole; Vfend; 137234-62-9; UK-109496; Voriconazol; Voriconazolum |
|---|---|
CAS 137234-62-9
(aR,bS)-a-(2,4-Difluorophenyl)-5-fluoro-b-methyl-a-(1H-1,2,4-triazol-1-ylmethyl)-4-pyrimideethanol
2R,3S-2-(2,4-difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1H-1,2,4-triazol-1-yl)butan-2-ol
Manufacturers' Codes: UK-109496
Trademarks: Vfend (Pfizer)
MF: C16H14F3N5O
MW: 349.31
Percent Composition: C 55.01%, H 4.04%, F 16.32%, N 20.05%, O 4.58%
Properties: mp 127°. [a]D25 -62° (c = 1 in methanol).
Melting point: mp 127°
Optical Rotation: [a]D25 -62° (c = 1 in methanol)
Therap-Cat: Antifungal (systemic)
|
Voriconazole (vor-i-KON-a-zole, brand name Vfend, Pfizer) is a triazole antifungal medication that is generally used to treat serious, invasive fungal infections. These are generally seen in patients who are immunocompromised, and include invasive candidiasis, invasive aspergillosis, and certain emerging fungal infections
1H NMR......... http://file.selleckchem.com/downloads/nmr/S144202-Voriconazole-HNMR-Selleck.pdf
1H NMR DMSO-d6, peak at 3.3 is HOD

m.p=134
1H-NMR (300 MHz, DMSO-d6) δ
(ppm):
9.04 (1H),
8.84 (1H),
8.23 (1H),
7.61 (1H),
7.28 (1H),
7.17 (1H),
6.91 (1H),
5.97 (1H),
4.80 (1H),
4.34 (1H),
3.93 (1H),
1.1 (3H).............US8263769
9.04 (1H),
8.84 (1H),
8.23 (1H),
7.61 (1H),
7.28 (1H),
7.17 (1H),
6.91 (1H),
5.97 (1H),
4.80 (1H),
4.34 (1H),
3.93 (1H),
1.1 (3H).............US8263769
13 C NMR
DMSO-d6

1H NMR PREDICT




13C NMR PREDICT



COSY PREDICT

HMBC PREDICT

HPLC



To date, only two methods for preparing voriconazole have been reported. One is based on a coupling reaction using an organic lithium salt, and the other, on Reformatsky-type coupling reaction.
For example, Korean Patent No. 1993-0011039 and European Patent No. 0,440,372 disclose a method shown in Reaction Scheme A for preparing the desired enantiomeric pair by a) adding an organic lithium derivative of 4-chloro-6-ethyl-5-fluoropyrimidine to 1-(2,4-difluorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethanone at −70° C.˜−50° C. to obtain an enantiomer mixture; and b) separating the desired enantiomer by chromatography.

PCT Publication No. WO 2006/065726 discloses a method shown in Reaction Scheme B for preparing the desired enantiomeric pair by repeating the procedure of Reaction Scheme A except for using a different solvent.

In order to solve the problems, as shown in Reaction Scheme C, Korean Patent Publication No. 1999-0036174 and U.S. Pat. No. 6,586,594 B1 disclose a method for preparing voriconazole by conducting Reformatsky-type reaction to enhance the stereoselectivity and yield, and then reductively removing the chlorine substituent in the presence of a palladium catalyst.

Further, the literature ([Organic Process Research & Development 2001, 5, 28-36], Pfizer Inc.) teaches that the chlorine substituent of the pyrimidine derivative adversely influences the coupling reaction pattern as shown in Reaction Scheme D and Table 1.

| TABLE 1 | ||||||
| Reformatsky-type reaction of compounds (VI, VII) and (IV) | ||||||
| Compound | Compound | Unreacted | Debrominated | Compound | Compound | |
| Pyrimidine | (VIII) (%) | (IX) (%) | pyrimidine (%) | pyrimidine (%) | (X) (%) | (XI) (%) |
| Compound | 47.5 | 24.0 | 0.0 | 15 | 4.3 | 9.2 |
| (VI) | ||||||
| Compound | 5.3 | 4.6 | 8.5 | 28 | 0.0 | 51.6 |
| (VII) | ||||||
MORE..................
In the synthesis of Voriconazole, it is
thought that two important steps are step i) of preparing the pyrimidine
derivative as an intermediate for use in the subsequent coupling
reaction with high yield and high purity, and step ii) of increasing
stereoselectivity in carrying out the coupling reaction between the
pyrimidine derivative and the ketone derivative to obtain the resultant
tertiary alcohol with high purity and high yield.
First, the pyrimidine derivative has been
prepared as depicted in the following Reaction Scheme 1 under reflux
without any solvent according to Korean Patent No. 1993-0011039 and EP
0440372. It is reported that the yield of pyrimidine derivative is as
low as 66%. However, the method of Reaction Scheme 1 is not suitable for
mass production owing to its severe reaction condition and low yield.
In addition, Korean Patent No. 10-0269048
and EP 0871625 disclose that the pyrimidine derivative is prepared via
the method of Reaction Scheme 1 in the presence of a solvent, and the
yield of the target product is 90%. However, in this case, there are
problems in that phosphoryl chloride used in an excessive amount is
hardly removed and the resultant product has low purity.

Meanwhile, Korean Unexamined Patent
Publication No. 10-2009-0014468 discloses a process for preparing
substituted thiopyrimidine derivatives by introducing a thiol group to a
pyrimidine derivative, as shown in the following Reaction Scheme 2, to
increase the purity of the pyrimidine derivative.

However, the above process is not amenable
to industrial mass production due to the increased number of steps as
compared to Reaction Scheme 1, the use of expensive thiol derivatives,
and the bad odor generated during the step using thiol. Next, Korean
Patent No. 1993-0011039 and EP 0440372 disclose processes for carrying
out a coupling reaction between pyrimidine derivatives and ketone
derivatives. Herein, as shown in the following Reaction Scheme 3, LDA
(lithium diisopropylamide), a strong base, or sodium
bis(trimethylsilyl)amide is used to perform the coupling reaction.

However, the above methods are problematic
in that they use highly explosive strong bases and require equipment
capable of cryogenic reaction. Above all, the methods provide very low
yield due to the low stereoselectivity and difficulty in separating
isomers, and thus are not amenable to mass production.
To overcome the above-mentioned problems,
Korean Patent No. 10-0269048 and EP 0871625 disclose a method by which
the stereoselectivity is increased through the Reformatsky-type coupling
reaction as depicted in the following Reaction Scheme 4, and
enantiomeric pairs (2R,3S/2S,3R) are separated in the form of their
hydrochloride salts via crystallization, thereby increasing the yield.

However, the method is problematic in that
it results in a relatively low yield of 65% despite a high ratio of the
enantiomeric pairs of 9:1 (2R,3S/2S,3R:2R,3R/2S,3S).
The method has another problem related to the removal of halo after the hydrochloride salts are treated with base.
EP 0069442 discloses a method for
preparing 1-(2,4-difluorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethanone, one
of the main intermediates of Voriconazole, according to the following
Reaction Scheme 5.

However, the above method provides a low yield of 40%.



..............................
Scheme E.

Example 7
Preparation of (2R,3S)-2-(2,4-difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1H-1,2,4-triazol-1-yl)butane-2-ol(voriconazole)
10 g of
(2R,3S)-2-(2,4-difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1H-1,2,4-triazol-1-yl)butane-2-ol
(R)-camsylate obtained in Example 6 was added to a mixture of 50 ml of
water and 50 ml of dichloromethane, and a 40% sodium hydroxide solution
was slowly added thereto to adjust the pH to 11˜12. The organic layer
was separated therefrom and dried over magnesium sulfate, and the
organic solvent was removed under a reduced pressure. The resulting
solution was crystallized with 18 ml of isopropanol, cooled to 0° C.,
stirred for 2 hours, and dried to obtain the white title compound (5.56
g, yield: 93%).
m.p=134
1H-NMR (300 MHz, DMSO-d6) δ
(ppm): 9.04 (1H), 8.84 (1H), 8.23 (1H), 7.61 (1H), 7.28 (1H), 7.17 (1H),
6.91 (1H), 5.97 (1H), 4.80 (1H), 4.34 (1H), 3.93 (1H), 1.1 (3H)
The optical purity of the compound obtained from HPLC analysis was >99.9%.
Comparative Example
Preparation of
(2R,3S)/(2S,3R)-(2R,3R)/(2S,3S)-3-(4-chloro-5-fluoropyrimidine-6-yl)-2-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)butane-2-ol
hydrochloride
5.29 g of zinc powder treated with 1N HCl and 0.26 g
of lead powder were added to 33.5 ml of tetrahydrofuran and stirred,
and 3.98 g of iodine dissolved in 10.6 ml of tetrahydrofuran was slowly
added thereto for 10 min while heating to 45° C. The resulting mixture
was cooled to 2° C., and a solution dissolving 3.53 g of
1-(2,4-difluorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethanone in 30 ml of
tetrahydrofuran, 5 g of 6-(1-bromo-ethyl)-4-chloro-5-fluoropyrimidine
and 0.32 g of iodine were slowly added thereto for 10 min. The obtained
mixture was heated to 25° C. and reacted for 1 hour.
4.67 g of glacial acetic acid and 12 ml of water
were added to the reaction solution, solid metal residue was filtered
out, and tetrahydrofuran was removed under a reduced pressure.
The resulting residue was extracted twice with 66 ml
of ethyl acetate, and the extract was successively washed with 4.67 g
of disodium ethylenediaminetetraacetate dehydrate dissolved in 12 ml of
water, and 30 ml of brine. The organic layer was concentrated to 40 ml
volume, and 0.86 g of HCl dissolved in 4.3 ml of isopropanol was added
thereto at 25° C.
The obtained crystal was filtrated, washed with 10
ml of ethyl acetate, and dried to obtain the title compound as a yellow
crystal (2.81 g, yield: 42%).
m.p=126˜130° C.
1H-NMR (300 MHz, DMSO-d6) δ (ppm): 8.84 (1H), 8.73 (1H), 7.93 (1H), 7.28 (1H), 7.20 (1H), 6.91 (1H), 4.82 (1H), 4.54 (1H), 3.93 (1H), 1.14 (3H)
The enantiomer ratio obtained from HPLC analysis of
the reaction solution by using an internal standard material was 10:1,
and 14.39% of unknown byproduct was formed. Further, the ratio of
(2R,3S)/(2S,3R)- and (2R,3R)/(2S,3S)-enantiomeric pair obtained from
HPLC analysis of the crystallized hydrochloride was 94.4%:4.8%.
.........................Patent WO2007132354A2

..................
http://www.google.co.in/patents/WO2009024214A1?cl=en

Example 18
(2R.3S)-2-(2.4-Difluorophenvn-3-(5-fluoropyrimidin-4-yl)-1 -d H-1.2.4-triazol-
1-yl)butan-2-ol (voriconazole), compound of Formula (\) To a solution of the racemic material as obtained in example 15 (1.28 g, 3.66 mmol) in acetone (29 ml) was added a solution of (1R)-10-camphorsulfonic acid (0.85 g, 3.66 mmol) in methanol (9.6 ml). The solvents were removed at reduced pressure, and the residue was dissolved in a mixture of acetone (10 ml) and methanol (2 ml). Crystals formed spontaneously after 3 h. Acetone (10 ml) was added, and the mixture was stirred overnight. The solid was isolated by filtration, washed with a small amount of acetone and dried. The solid was dissolved in a mixture of acetone (14 ml) and methanol (4 ml) at reflux. The solution was cooled to rt and stirred for 90 min. Isolation of the precipitate formed by filtration, washing with acetone and drying afforded 0.72 g of the acid addition salt. 0.70 g of the solid material was taken up in dichloromethane (10 ml) and water (10 ml), and the pH was adjusted to 11 by addition of aqueous sodium hydroxide (15% sol.). The layers were separated, and the aqueous layer was extracted with dichloromethane (5 ml). The combined organic layers were washed with water (3 x 10 ml) and brine, and dried (sodium sulfate). Concentration at reduced pressure afforded 0.36 g (28% yield, 56% of the available enantiomer) of voriconazole as a white crystalline solid. Purity, HPLC: 99.8% (RT=4.91 min). Mp. 122.6 0C (Lit. 134 0C). MS, m/z (% rel. int.): 224.0 (27), 350.1 (100), 391.0 (10). 1H NMR (600 MHz, DMSO-d6): δ 9.02 (1H, d, J 3.0 Hz)1 8.83 (1 H1 d, J 1.8 Hz), 8.21 (1 H1 s), 7.59 (1 H1 s), 7.24 (1H1 ddd, J 7.0 Hz, J 9.0 Hz, J 9.0 Hz), 7.16 (1H, ddd, J 2.4 Hz, J 9.0 Hz, J 11.8 Hz), 6.89 (1H, ddd, J 2.4 Hz, J 8.4 Hz, J 8.4 Hz), 5.95 (1 H, s), 4.77 (1 H, d, J 14.4 Hz), 4.31 (1H, d, J 14.4 Hz), 3.90 (1 H, q, J 7.0 Hz), 1.08 (3H1 d, J 7.0 Hz).
Example 19
(2R.3S)-2-(2.4-Difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1 -d H-1.2.4-triazol-
1 -yl)butan-2-ol (voriconazole), compound of Formula (I) (2R,3S)-2-(2,4-Difluorophenyl)-3-[5-fluoropyrimidin-4-yl]-1-(1 H-1 ,2,4-triazol- 1-yl)butan-2- ol (R)-10-camphorsulfonate (656 g, 1.13 mol) was parted between dichloromethane (3 L) and water (3 L), and the pH of the aqueous phase was slowly adjusted to pH 12.3 with aqueous sodium hydroxide (40% w/v, 130 mL). The phases were separated, and the aqueous layer was extracted with dichloromethane (1 L). The combined organic layers were washed with water (3 x 1.5 L) and filtered. The solvent was changed to isopropanol (1.3 L) via gradual addition/evaporation at reduced pressure. After stirring for 3 h at 20 0C, the temperature was lowered to -5 0C. The mixture was stirred for another 1.5 h, and the product was isolated by suction filtration, washed with isopropanol (0.3 L) and dried at 20 0C and vacuum for 2.5 days. 285 g (72% yield) of the title compound was obtained as a white solid. Purity, HPLC: 99.8%. Optical purity, HPLC: >99.9%. 1H NMR (300 MHz, DMSO-d6): δ 9.05 (1H1 d, J 3.0 Hz), 8.86 (1H1 d, J 1.8 Hz), 8.24 (1H, s), 7.62 (1H, s), 7.24 (2H, m), 6.92 (1H, dt, J 2.1, 8.0 Hz), 5.99 (1H, s), 4.81 (1H1 d, J 14.1 Hz), 4.34 (1H1 d, J 14.4 Hz), 3.93 (1H, q, J 7.2 Hz), 1.11 (3H1 d, J 6.9 Hz). 13C NMR spectrum and IR spectrum for the isolated compound are attached.


......................
PAPER
PAPER
J. Org. Chem., 2013, 78 (22), pp 11396–11403
DOI: 10.1021/jo4019528
..........................
Org. Proc. Res. Dev., 2001, 5 (1), pp 28–36
DOI: 10.1021/op0000879
(2R,3S)-2-(2,4-Difluorophenyl)-3-(5-fluoro-4-pyrimidinyl)-1-(1H-1,2,4-triazol-1-yl)-2-butanol (1). ...............to provide the title compound as a white solid (7.6 g, 40% mass yield or 80% of available enantiomer), mp 134 °C
1H NMR (DMSO-d6)
δ 1.1 (d, 3H), 3.93 (q, 1H), 4.34 (d, 1H), 4.80 (d, 1H), 5.97 (bs, 1H),
6.91 (ddd, 1H), 7.17 (ddd, 1H), 7.28 (ddd, 1H), 7.61 (s, 1H), 8.23 (s,
1H), 8.84 (s, 1H), 9.04 (s, 1H) ppm.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6586594 | 26 Jul 1996 | 1 Jul 2003 | Pfizer, Inc. | Preparation of triazoles by organometallic addition to ketones and intermediates therefor |
| CN1488630A | 8 Oct 2002 | 14 Apr 2004 | 张文更 | Method for preparing triazole antifungal agent |
| CN1814597A | 9 Dec 2005 | 9 Aug 2006 | 北京丰德医药科技有限公司 | New method for preparing voriconazole |
| EP0440372A1 | 24 Jan 1991 | 7 Aug 1991 | Pfizer Limited | Triazole antifungal agents |
| GB2452049A | Title not available | |||
| WO1993007139A1 | 1 Oct 1992 | 15 Apr 1993 | Pfizer Ltd | Triazole antifungal agents |
| WO1997006160A1 | 26 Jul 1996 | 20 Feb 1997 | Michael Butters | Preparation of triazoles by organometallic addition to ketones and intermediates therefor |
| WO2006065726A2 | 13 Dec 2005 | 22 Jun 2006 | Reddys Lab Ltd Dr | Process for preparing voriconazole |
| WO2007013096A1 | 26 Jun 2006 | 1 Feb 2007 | Msn Lab Ltd | Improved process for the preparation of 2r, 3s-2-(2,4-difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1h-1,2,4-triazol-1-yl) butan-2-ol (voriconazole) |
| WO2007132354A2 | 29 Jan 2007 | 22 Nov 2007 | Medichem Sa | Process for preparing voriconazole, new polymorphic form of intermediate thereof, and uses thereof |
| WO2009024214A1 * | 10 Jul 2008 | 26 Feb 2009 | Axellia Pharmaceuticals Aps | Process for the production of voriconazole |
| WO2009084029A2 | 2 Dec 2008 | 9 Jul 2009 | Venkatesh Bhingolikar | Improved process for the preparation of (2r,3s)-2-(2,4- difluqrophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1h-1,2,4-triazol-1-yl) butan-2-ol |
| US8575344 * | 1 Feb 2011 | 5 Nov 2013 | Dongkook Pharmaceutical Co., Ltd. | Process for preparing voriconazole by using new intermediates |
| US20130005973 * | 1 Feb 2011 | 3 Jan 2013 | Dongkook Pharmaceutical Co., Ltd. | Process for preparing voriconazole by using new intermediates |
| |||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Butters et al., "Process Development of Voriconazole: A Novel Broad-Spectrum Triazole Antifungal Agent," Organic Process Research & Development, 2001, vol. 5, pp. 28-36. | ||||||||||||||||||||||||||||||||||||
References:
Ergosterol biosynthesis inhibitor. Prepn: S. J. Ray, K. Richardson, EP 440372; eidem, US 5278175 (1991, 1994 both to Pfizer); R. P. Dickinson et al., Bioorg. Med. Chem. Lett. 6, 2031 (1996).
Mechanism of action: H. Sanati et al.,Antimicrob. Agents Chemother. 41, 2492 (1997). In vitro antifungal spectrum: F. Marco et al., ibid. 42, 161 (1998).
HPLC determn in plasma: R. Gage, D. A. Stopher, J. Pharm. Biomed. Anal. 17, 1449 (1998).
Review of pharmacology and clinical development: P. E. Verweij et al., Curr. Opin. Anti-Infect. Invest. Drugs 1, 361-372 (1999); J. A. Sabo, S. M. Abdel-Rahman, Ann. Pharmacother. 34, 1032-1043 (2000).
Clinical pharmacokinetics: L. Purkins et al., Antimicrob. Agents Chemother. 46, 2546 (2002).
Clinical comparison with amphotericin B: T. J. Walsh et al., N. Engl. J. Med. 346, 225 (2002).
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
(2R,3S)-2-(2,4-Difluorophenyl)-3-(5-fluoropyrimidin-4-yl)-1-(1H-1,2,4-triazol-1-yl)butan-2-ol
|
|
| Clinical data | |
| Trade names | VFEND |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a605022 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
IV, oral |
| Pharmacokinetic data | |
| Bioavailability | 96% |
| Protein binding | 58% |
| Metabolism | Hepatic cytochrome P450 enzymes CYP2C19, CYP2C9, CYP3A4 |
| Biological half-life | Dose-dependent |
| Identifiers | |
| CAS Registry Number | 137234-62-9 |
| ATC code | J02AC03 |
| PubChem | CID: 71616 |
| DrugBank | DB00582 |
| ChemSpider | 64684 |
| UNII | JFU09I87TR |
| KEGG | D00578 |
| ChEBI | CHEBI:10023 |
| ChEMBL | CHEMBL638 |
| Chemical data | |
| Formula | C16H14F3N5O |
| Molecular mass | 349.311 g/mol |
11
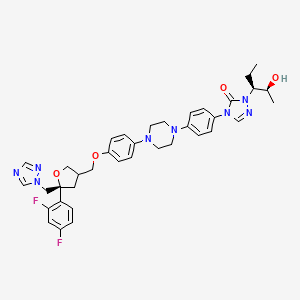
POSACONAZOLE


Posaconazole 泊沙康唑 , بوساكونازول , Позаконазол
Sch-
56592
4-[4-[4-[4-[[(5R)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)oxolan-3-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-2-[(2S,3S)-2-hydroxypentan-3-yl]-1,2,4-triazol-3-one
- Noxafil
- SCH 56592
U.S. Patents 5,661,151; 5,703,079; and 6,958,337.
Therap-Cat: Antifungal.
CAS 171228-49-2
Molecular Formula: C37H42F2N8O4
Molecular Weight: 700.78
CAS Name: 2,5-Anhydro-1,3,4-trideoxy-2-C-(2,4-difluorophenyl)-4-[[4-[4-[4-[1-[(1S,2S)-1-ethyl-2-hydroxypropyl]-1,5-dihydro-5-oxo-4H-1,2,4-triazol-4-yl]phenyl]-1-piperazinyl]phenoxy]methyl]-1-(1H-1,2,4-triazol-1-yl)-D-threo-pentitol
Additional Names: (3R-cis)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin-1-yl]phenyl]-2-[1(S)-ethyl-2(S)-hydroxypropyl]-3,4-dihydro-2H-1,2,4-triazol-3-one
Syn..........Dominic De Souza, "PREPARATION OF POSACONAZOLE INTERMEDIATES." U.S. Patent US20130203994, issued August 08, 2013.
Percent Composition: C 63.41%, H 6.04%, F 5.42%, N 15.99%, O 9.13%
Melting Point
170-172 deg C
O'Neil,
M.J. (ed.). The Merck Index - An Encyclopedia of Chemicals, Drugs, and
Biologicals. 13th Edition, Whitehouse Station, NJ: Merck and Co., Inc.,
2001., p. 1365
-
In water, 0.027 mg/L at 25 deg C (est)
US EPA; Estimation Program Interface (EPI) Suite. Ver.3.12. Nov 30, 2004. Available from, as of Dec 19, 2005:http://www.epa.gov/oppt/exposure/pubs/episuitedl.htm
US5661151 EXP Jul 19, 2019 PRODUCT PATENT
US 5703079 EXP Aug 26, 2014
US8410077 EXPMar 13, 2029
US9023790 EXPJul 4, 2031
US 6958337 EXP Oct 5, 2018
US 8263600 EXPApr 1, 2022
Posaconazole is a triazole antifungal drug[1][2] marketed in the United States, the European Union, and in other countries by Schering-Plough under the trade name Noxafil. In Canada, posaconazole is marketed by Schering-Plough under the trade name Posanol.
Pharmacology
Mode of action
Posaconazole works by disrupting the close packing of acyl chains of phospholipids,
impairing the functions of certain membrane-bound enzyme systems such
as ATPase and enzymes of the electron transport system, thus inhibiting
growth of the fungi. It does this by blocking the synthesis of ergosterol by inhibiting of the enzymelanosterol 14α-demethylase
and accumulation of methylated sterol precursors. Posaconazole is
significantly more potent at inhibiting 14-alpha demethylase than itraconazole.[3][4][5]
Microbiology
- Candida spp.
- Aspergillus spp.
- Zygomycetes spp.
Pharmacokinetics
Posaconazole is absorbed within three to five hours. It is predominately eliminated through the liver, and has a half life of about 35 hours. Oral administration of posaconazole taken with a high-fat meal exceeds 90% bioavailabilityand increases the concentration by four times compared to fasting state.[6][7]
Clinical use
It is used to treat invasive infections by Candida species,[8] Mucor, and Aspergillus species[9] in severelyimmunocompromised patients.
Clinical evidence for its utility in treatment of invasive disease caused by Fusarium species (fusariosis) is limited.[10]
Two studies suggest posaconazole may be superior to other triazoles, such as fluconazole or itraconazole, in the prevention of invasive fungal infections, although it may cause more serious side effects.[11][12]
There
is also some indication that posaconazole may be the most effective
treatment for both chronic and acute Chagas disease, showing much better
efficacy than benznidazole.[13] Schering-Plough
is currently recruiting participants for a phase 2 clinical trial in
Argentina to test its efficacy against asymptomatic, chronic Chagas.[14]
SYNTHESIS



................
http://www.google.com/patents/WO2013042138A2?cl=en
HPLC Method of Analysis for Posaconazole:
Posaconazole is analyzed by HPLC using the following conditions: Apparatus: A liquid chromatographic system is to be equipped with variable wavelength UV-detector; Column: Grace Alltima CI 8, 150 x 4.6mm 3μηι or equivalent; Flow rate: 1.0 ml/min; Wavelength: 210 nm; Column Temperature: 28°C; Injection volume: 10
The particle size distribution of posaconazole compound of formual-1 is measured using the following conditions:
Instrument: Malvern Master sizer 2000; Measuring range: 0.02 to 2000 μπι; Wet sample: Hydro 2000S; Dispersant: Water; Absorption Index: 0; Refractive Index of water: 1.330; Refractive Index of particle: 1.500; Stirrer speed: 2500 rpm; Obscuration range: 10-20%; Sensitivity: Normal; Measurement time: 12 seconds; Background time: 12 seconds; Internal sonication: 3 minutes; (Tip displacement-70%); Measurement repeat: 3 times at zero second interval.
HPLC Method of Analysis for Benzylated Posaconazole:
Benzylated posaconazole is analyzed by HPLC using the following conditions:
Apparatus: A liquid chromatographic system is to be equipped with variable wavelength UV-detector; Column: X-bridge C18, 50X 4.6mm, 3.5um (or) equivalent; Flow rate: 0.8 ml/min; Wavelength: 210 nm; Column Temperature: 40°C; Injection volume: 5 μΐ,; Run time: 35 min; Diluent: Water: Acetonitrile (40:60) v/v; Needle wash: Water: Acetonitrile (40:60) v/v; Elution: Gradient; Mobile phase-A: Buffer Acetonitrile (90: 10) v/v; Mobile phase-B: Acetonitrile: water (90:10) v/v; Buffer: 1.74 grams of potassium hydrogen phosphate dibasic (anhydrous) in 1000 ml of Milli-Q- Water. Adjust its pH to 6.5 with diluted orthophosphoric acid and filtered through 0.22μπι Nylon membrane filter paper and sonicate to degas it. PXRD analysis of crystalline triazole antifungal compound of formula- 1 was carried out using BRUKER/AXS X-Ray diffractometer using Cu Ka radiation of wavelength 1.5406 A° and continuous scan speed of 0.03°/min.
RS/OVI analysis of amorphous posaconazole is carried out on Agilent GC-6850 series-2 with Flame Ionization detector, column AP vac, flow 2 psi and load is 1 μΐ, detector temperature is 260°C and carrier gas is helium.
The process of the present invention is schematically represented as below:
Scheme-I

Scheme-II:

HO N N- NH,
Formula-18

Patent WO2013042138A2



Example-16: Preparation of Posaconazole (Formula- 1)
5N hydrochloric acid (72 ml) and 10% Pd-C (10 g) were added to a solution of 4- (4_(4-(4-(((3Κ,5Κ)-5-(( i H- 1 ,2,4-triazol- 1 -yl)methyl)-5-(2,4-difluorophenyl)tetrahydro furan-3-yl)methoxy)phenyl)piperazin-l-yl)phenyl)-l-((2S,3R)-2-(benzyloxy)pentan-3- yl)-lH-l,2,4-triazol-5(4H)-one compound of formula-21 (42 g) in methanol (420 ml). The reaction mixture was hydrogenated for 5 hours under a hydrogen gas pressure of 4-5 kg/cm2 at 50°. After completion of reaction, the catalyst was filtered off and washed with methanol. pH of the filtrate was adjusted to ~7.0 using 4N sodium hydroxide. Water was added to the reaction mixture and stirred for 2 hours at 25-35°C. Filtered the separated solid and washed with water. The obtained solid was dissolved in acetone (320 ml)and stirred at reflux temperature for 30 minutes. Filtered the undissolved product and added water to the filtrate and stirred the reaction mixture for 4 hours at 25-35°C. Filtered the separated solid and washed with water. Further the solid was recrystallized from isopropyl alcohol to get the title compound. Purity by HPLC: 99.85%; Yield: 75.0%: Chiral purity by HPLC: 99.82%.
......................
Posaconazole, SCH-56592, Noxafil
EP 0736030; JP 1997500658; US 5661151; US 5703079; WO 9517407
Synthesis
of intermediate (XX):
The reaction of 2-chloro-2',4'-difluoroacetophenone (I) with sodium
acetate and NaI in DMF gives 2-acetoxy-2',4'-difluoroacetophenone (II),
which by methylenation with methyltriphenylphosphonium bromide and
sodium bis(trimethylsilyl)amide in THF yields
2-(2,4-difluorophenyl)-2-propen-1-ol acetate ester (III). The hydrolysis
of (III) with KOH in dioxane/water affords the corresponding alcohol
(IV), which is regioselectively epoxidized with titanium
tetraisopropoxide and L-(+)-diethyl tartrate in dichloromethane to
(S)-(-)-2-(2,4-difluorophenyl)oxirane-2-methanol (V). The reaction of
(V) with 1,2,4-triazole (VI) in DMF affords
(R)-2-(2,4-difluorophenyl)-3-(1,2,4-triazol-1-yl)propane-1,2-diol (VII),
which is selectively mesylated with methanesulfonyl chloride and
triethylamine to the monomesylate (VIII). The cyclization of (VIII) with
NaH in DMF gives the oxirane (IX), which is condensed with diethyl
malonate (X) by means of NaH in DMSO to yield a mixture of (5R-cis)- and
(5R-trans)-5-(2,4-difluorophenyl)-2-oxo-5-(1,2,4-triazol-1-ylmethyl)
tetrahydrofuran-3-carboxylic acid ethyl ester (XI). The reduction of
(XI) with NaBH4 and LiCl in ethanol affords
(R)-4-(2,4-difluorophenyl)-2-(hydroxymethyl)-5-(1,2,4-triazol-1-yl)
pentane-1,4-diol (XII), which is selectively tosylated with tosyl
chloride and triethylamine in THF to the bistosylate (XIII). The
cyclization of (XIII) by means of NaH in refluxing toluene gives
(5R-cis)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)
tetrahydrofuran-3-methanol tosylate ester (XIV). The reaction of (XIV)
with 1-(4-hydroxyphenyl)-4-(4-nitrophenyl)piperazine (XV) to obtain
compound (XVI), and the following reaction sequence (XVI) to (XVII) to
(XVIII) to (XIX) to
(5R-cis)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin-1-yl]phenyl-3,4-dihydro-2H-1,2,4-triazol-3-one
(XX) has been performed according to J Med Chem 1984, 27: 894-900.
1. An improved process for the preparation of (3S,5R)-5-(2,4-difluorophi
(iodomethyl)tetrahydrofuran-3-ca boxylic acid compound of formula-7,

Forrnula-7
comprising of the following steps:
a) Reacting 4-(2,4-difluorophen ent-4-enoic acid compound of formula-2


Formula-2
with (R)-4-phenyloxazolidin-2-one compound of formula-3

Formula-3
in presence of a suitable activating
agent and a suitable base in a suitable solvent to provide
(R)-3-(4-(2,4-difluorophenyl)pent-4-enoyl)-4-phenyloxazolidin-2-one
compound of formula-4,


Formula-4
b) hydroxymethylating the compound of
formula-4 with 1,3,5-trioxane in presence of a base and a catalyst in a
suitable solvent to provide (R)-3-((S)-4-(2,4-
difluorophenyl)-2-(hydroxymethyl)pent-4-enoyl)-4-phenyloxazolidin-2-one
compound of formula-5,


Formula-5
c) cyclizing the compound of formula-5
in-situ in presence of iodine and a suitable base in a suitable solvent
to provide (R)-3-((3S,5R)-5-(2,4-difluorophenyl)-5-
(iodomethyl)tetrahydrofuran-3 -carbonyl)-4-phenyloxazolidin-2-one
compound of formula-6,

Formula-6
d) hydrolyzing the compound of formula-6
in presence of a suitable aqueous base and hydrogen peroxide in a
suitable solvent to provide (3S,5R)-5-(2,4-
difluorophenyl)-5-(iodomethyl)tetrahydrofuran-3-carboxylic acid compound
of formula-7.
formula-7,



Formula-7
b) reducing the compound of formula-7
with a suitable reducing agent in a suitable solvent to provide
((3R,5R)-5-(2,4-difluorophenyl)-5-(iodomethyl)tetrahydro
furan-3-yl)methanol compound of formula-8,

Formula-8
Example-16: Preparation of Posaconazole (Formula- 1)
5N hydrochloric acid (72 ml) and 10% Pd-C (10 g) were added to a solution of 4- (4_(4-(4-(((3Κ,5Κ)-5-(( i H- 1 ,2,4-triazol- 1 -yl)methyl)-5-(2,4-difluorophenyl)tetrahydro furan-3-yl)methoxy)phenyl)piperazin-l-yl)phenyl)-l-((2S,3R)-2-(benzyloxy)pentan-3- yl)-lH-l,2,4-triazol-5(4H)-one compound of formula-21 (42 g) in methanol (420 ml). The reaction mixture was hydrogenated for 5 hours under a hydrogen gas pressure of 4-5 kg/cm2 at 50°. After completion of reaction, the catalyst was filtered off and washed with methanol. pH of the filtrate was adjusted to ~7.0 using 4N sodium hydroxide. Water was added to the reaction mixture and stirred for 2 hours at 25-35°C. Filtered the separated solid and washed with water. The obtained solid was dissolved in acetone (320 ml)and stirred at reflux temperature for 30 minutes. Filtered the undissolved product and added water to the filtrate and stirred the reaction mixture for 4 hours at 25-35°C. Filtered the separated solid and washed with water. Further the solid was recrystallized from isopropyl alcohol to get the title compound. Purity by HPLC: 99.85%; Yield: 75.0%: Chiral purity by HPLC: 99.82%.
......................
Posaconazole, SCH-56592, Noxafil
EP 0736030; JP 1997500658; US 5661151; US 5703079; WO 9517407

The bromosulfonate (XXI) has been obtained as follows: (S)-Lactic acid methyl ester (XXII) has been protected as its benzyloxymethyl ether (XXIII) according to Tetrahedron Lett 1980, 21: 1035. The reduction of (XXIII) with DIBAL yields the corresponding aldehyde (XXIV), which by a Grignard reaction with ethylmagnesium bromide in THF and chromatographic separation of the diastereoisomers (SiO2, hexane/ethyl acetate) affords 2(S)-(benzyloxymethoxy)-3(R)-pentanol (XXV). Finally, this compound is sulfonated to (XXI) with 4-bromobenzenesulfonyl chloride. Finally, compound (XX) is condensed with 2(S)-(benzyloxymethoxy)-3(R)-pentanol 4-bromobenzenesulfonate ester (XXI) by means of cesium carbonate in DMF, and deprotected with 6N HCl.
.....................
WO 9633178
A new synthesis of Sch-56592 has been described: The reaction of (S)-ethyl lactate (I) with pyrrolidine (II) gives 1-[(S)-lactoyl]pyrrolidine (III), which is benzylated as usual with benzyl chloride yielding the benzyl ether (IV). The reaction of (IV) with ethylmagnesium bromide in THF affords 2(S)-benzyloxy-3-pentanone (V), which is reduced with LiBH4 in dimethoxyethane giving 2(S)-benzyloxy-3(RS)-pentanol (VI). The reaction of (VI) with 4-chlorobenzenesulfonyl chloride (VII) yields the corresponding sulfonate (VIII), which is treated with hydrazine in ethanol to afford a diastereomeric mixture of hydrazines that is resolved with L-dibenzoyltartaric acid giving the (S,S)-enantiomer (IX). The formylation of (IX) with refluxing ethyl formate yields the chiral formyl hydrazide (X), which is cyclized with N-[4-[4-[4-(trimethylsilyloxy)phenyl]piperazin-1-yl]phenyl]carbamic acid phenyl ester (XI) affording the triazolone (XII). Finally, this compound is condensed with the chiral tetrahydrofuran derivative (XIII) by means of NaOH in DMSO, and debenzylated by hydrogenation with H2 over Pd/C in formic acid
.....................
\35th Intersci Conf Antimicrob Agents Chemother (Sept 17-20, San Francisco) 1995,Abst F83.

2) The 2-(2,4-difluorophenyl)-2-propen-1-ol (IV) is converted into the corresponding propenyl bromide (XXVI), which is condensed with diethyl malonate (X) to afford the malonyl derivative (XXVII). The reduction of (XXVII) with NaBH4 and LiCl yields the 1,3-propanediol derivative (XXVIII), which is enantioselectively acetylated with vinyl acetate and Novozyme 435 in acetonitrile yielding the isomeric (S)-monoacetate (XXIX). The cyclization of (XXIX) with iodine and NaHCO3 in acetonitrile affords (5R-cis)-5-(2,4-difluorophenyl)-5-(iodomethyl)tetrahydrofuran-3-methanol acetate ester (XXX), which is condensed with sodium 1,2,4-triazole (XXXI) to give (5R-cis)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-methanol acetate ester (XXXII). The hydrolysis of (XXXII) with NaOH yields the corresponding methanol (XXXIII), which is finally tosylated to the tosyl ester (XIV), already obtained previously.
.........................
35th Intersci Conf Antimicrob Agents Chemother (Sept 17-20, San Francisco) 1995,Abst F61

3) The Friedel Crafts condensation of m-difluorobenzene (XXXIV) with succinic anhydride (XXXV) gives 4-(2,4-difluorophenyl)-4-oxobutyric acid (XXXVI), which is converted by a Wittig reaction into 4-(2,4-difluorophenyl)-4-pentenoic acid (XXXVII) and subsequently into its acyl chloride (XXXVIII). The condensation of (XXXVIII) with 4(R)-benzyloxazolidin-2-one (XXXIX) gives the acyl oxazolidinone (XL), which is regioselectively hydroxymethylated with 1,3,5-trioxane and TiCl4 to afford 4(R)-benzyl-3-[4-(2,4-difluorophenyl)-3(S)-(hydroxymethyl)-4-pentenoyl] oxazolidin-2-one (XLI). The cyclization of (XLI) with iodine and pyridine yields the tetrahydrofuran derivative (XLII), which is reduced with LiBH4 to (5R-cis)-5-(2,4-difluoromethyl)-5-(iodomethyl)tetrahydrofuran-3-methanol (XLIII). Finally, this compound is condensed with sodium 1,2,4-triazole (XXXI) to afford (5R-cis)-5-(2,4-difluoromethyl)-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-methanol (XXXIII), already obtained in Scheme 22656201c.
.................
J Label Compd Radiopharm 1998,41(8),731
The synthesis of [3H]-SCH-51048 has been described: The tritiation of phenol (I) with tritiated heptafluorobutyric acid at 115 C gives the polytritiated intermediate (II), which is then condensed with the chiral tosylate (III) by means of NaOH in DMSO affording labeled SCH-51048.
......................

The synthesis of [14C]-SCH-56592 has been described: The cyclization of semicarbazide (I) with [14C]-formamidine (II) in hot 2-methoxyethanol gives the triazolone (III), which is condensed with the sulfonate (IV) by means of Cs2CO3 in hot DMF to yield the alkylated triazolone (V). Finally, this compound is deprotected by hydrogenation with formic acid over Pd/C in hot methanol to afford labeled SCH-56592.
..................
Tetrahedron Lett 2002,43(18),3359

The condensation of 4-chlorophenylsulfonate (I) with 4-bromophenol (II) by means of K2CO3 in hot DMF gives the aryl ether (III), which is condensed with piperazine (IV) by means of Pdo to yield the monosubstituted piperazine (V). Finally, this compound is condensed with the 4-bromophenyltriazolone (VI) by means of K2CO3 in hot DMSO to afford the target disubstituted piperazine

Alternatively, the condensation of with the 4-bromophenyltriazolone (VI) with piperazine (IV) by means of Pd2(dba)3, BINAP and t-BuONa in hot toluene gives the monosubstituted piperazine (VII), which is then condensed with the already reported aryl ether (III) by means of Pd2(dba)3, BINAP and t-BuONa in hot toluene, and debenzylated with Pd/C and formic acid to afford the target disubstituted piperazine.

The intermediate 4-[4-(4-aminophenyl)piperazin-1-yl]phenol (VIII) has been obtained by several related ways: 1.- The condensation of 4-bromonitrobenzene (I) with piperazine (II) gives 1-(4-nitrophenyl)piperazine (III), which is condensed with 4-bromoanisole (IV) by means of Pdo to yield 1-(4-methoxyphenyl)-4-(4-nitrophenyl)piperazine (V). Alternatively, (V) can also be obtained by condensation of (III) with 4-methoxyphenylboronic acid (VI) by means of Cu(OAc)2 in DMSO. The demethylation of (V) with HBr yields 4-[4-(4-nitrophenyl)piperazin-1-yl]phenol (VII), which is finally reduced with H2 over Pd/C to afford the target 4-[4-(4-aminophenyl)piperazin-1-yl]phenol (VIII) intermediate (see Synthline, scheme no. 22656202a, intermediate (XIV)). 2.- The condensation of piperazine (II) with 4-bromoanisole (IV) by means of Pdo gives 1-(4-methoxyphenyl)piperazine (IX), which is condensed with 4-bromonitrobenzene (I) by means of K2CO3 and tetrabutylammonium iodide (TBAI) in hot DMSO to yield intermediate 1-(4-methoxyphenyl)-4-(4-nitrophenyl)piperazine (V), already reported. 3.- The condensation of piperazine (II) with 4-(benzyloxy)phenyl bromide (X) by means of Pdo gives 1-(4-benzyloxyphenyl)piperazine (XI), which is condensed with 4-bromonitrobenzene (I) by means of K2CO3 and TBAI in hot DMSO to yield 1-(4-benzyloxyphenyl)-4-(4-nitrophenyl)piperazine (XII). The nitro group of (XII) is reduced by means of H2 (50 psi) over Pd/C in wet THF at 50? C to afford 4-[4-(4-benzyloxyphenyl)piperazin-1-yl]aniline (XIII), which is finally debenzylated with H2 (80 psi) over Pd/C in wet THF at 70? C or other drastic conditions to afford the target 4-[4-(4-aminophenyl)piperazin-1-yl]phenol (VIII) intermediate (see Synthline, scheme no. 22656202a, intermediate (XIV)). Alternatively, 1-(4-benzyloxyphenyl)-4-(4-nitrophenyl)piperazine (XII) can also be reduced directly to the target intermediate (VIII) with H2 over Pd/C under a variety of drastic conditions.
Literature References:
Orally active triazole antifungal. Prepn: A. K. Saksena et al., WO 9517407; eidem, US 5661151 (1995, 1997 both to Schering); eidem, Tetrahedron Lett. 37, 5657 (1996).
Comparative antifungal spectrum: A. Cacciapuoti et al., Antimicrob. Agents Chemother. 44, 2017 (2000). Pharmacokinetics, safety and tolerability: R. Courtney et al., ibid. 47, 2788 (2003).
HPLC determn in serum: H. Kim et al., J. Chromatogr. B 738, 93 (2000).
Review of development: A. K. Saksena et al. inAnti-Infectives: Recent Advances in Chemistry and Structure Activity Relationships (Royal Soc. Chem., Cambridge, 1997) pp 180-199; and clinical efficacy in fungal infections: R. Herbrecht, Int. J. Clin. Pract. 58, 612-624 (2004).
References
- ^ Schiller DS, Fung HB (September 2007)."Posaconazole: an extended-spectrum triazole antifungal agent". Clin Ther 29 (9): 1862–86.doi:10.1016/j.clinthera.2007.09.015.PMID 18035188.
- ^ Rachwalski EJ, Wieczorkiewicz JT, Scheetz MH (October 2008). "Posaconazole: an oral triazole with an extended spectrum of activity". Ann Pharmacother 42(10): 1429–38. doi:10.1345/aph.1L005.PMID 18713852.
- ^ a b Brunton L, Lazo J, Parker K. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. San Francisco: McGraw-Hill; 2006. ISBN 978-0-07-142280-2
- ^ "Clinical Pharmacology Pasoconazole". Retrieved18 February 2010.
- ^ "Daily Med, Product Information Noxafil". Retrieved18 February 2010.
- ^ a b Dodds Ashley, Elizabeth; Perfect, John (October 13, 2009). "Pharmacology of azoles". Retrieved18 February 2010.
- ^ "Drugs at FDA: Noxafil" (PDF). Retrieved18 February 2010.
- ^ Li X, Brown N, Chau AS et al. (January 2004)."Changes in susceptibility to posaconazole in clinical isolates of Candida albicans". J. Antimicrob. Chemother. 53 (1): 74–80. doi:10.1093/jac/dkh027.PMID 14657086.
- ^ Walsh TJ, Raad I, Patterson TF et al. (January 2007)."Treatment of invasive aspergillosis with posaconazole in patients who are refractory to or intolerant of conventional therapy: an externally controlled trial".Clin. Infect. Dis. 44 (1): 2–12. doi:10.1086/508774.PMID 17143808.
- Raad I, Hachem R, Herbrecht R et al. (2006)."Posaconazole as salvage treatment for invasive fusariosis in patients with underlying hematologic malignancy and other conditions". Clin Infect Dis 42(10): 1398–1403.
- Cornely O, Maertens J, Winston D, Perfect J, Ullmann A, Walsh T, Helfgott D, Holowiecki J, Stockelberg D, Goh Y, Petrini M, Hardalo C, Suresh R, Angulo-Gonzalez D (2007). "Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia". N Engl J Med356 (4): 348–59. doi:10.1056/NEJMoa061094.PMID 17251531.
- Ullmann A, Lipton J, Vesole D, Chandrasekar P, Langston A, Tarantolo S, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S (2007). "Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease". N Engl J Med 356 (4): 335–47. doi:10.1056/NEJMoa061098.PMID 17251530.
- "Am J Trop Med Hyg April 2010 vol. 82 no. 4"
- "A Study of the Use of Oral Posaconazole (POS) in the Treatment of Asymptomatic Chronic Chagas Disease (P05267 AM1) (STOP CHAGAS)"
Noxafil is an azole antifungal agent available
as concentrated solution to be diluted before intravenous
administration, delayed-release tablet, or suspension for oral
administration.
Posaconazole
is designated chemically as 4-[4-[4-[4-[[
(3R,5R)-5-(2,4-difluorophenyl)tetrahydro-5(1H-1,2,4-triazol-1-ylmethyl)-3-furanyl]methoxy]phenyl]-1-piperazinyl]phenyl]-2-[(1S,2S)-1-ethyl-2hydroxypropyl]-2,4-dihydro-3H-1,2,4-triazol-3-one
with an empirical formula of C37H42F2N8O4 and a molecular weight of 700.8. The chemical structure is:
Posaconazole is a white powder with a low aqueous solubility.

Noxafil
injection is available as a clear colorless to yellow, sterile liquid
essentially free of foreign matter. Each vial contains 300 mg of
posaconazole and the following inactive ingredients: 6.68 g Betadex
Sulfobutyl Ether Sodium (SBECD), 0.003 g edetate disodium, hydrochloric
acid and sodium hydroxide to adjust the pH to 2.6, and water for
injection.
Noxafil
delayed-release tablet is a yellow, coated, oblong tablet containing
100 mg of posaconazole. Each delayed-release tablet contains the
inactive ingredients: hypromellose acetate succinate, microcrystalline
cellulose, hydroxypropylcellulose, silicon dioxide, croscarmellose
sodium, magnesium stearate, and Opadry® II Yellow (consists of the
following ingredients: polyvinyl alcohol partially hydrolyzed,
Macrogol/PEG 3350, titanium dioxide, talc, and iron oxide yellow).
Noxafil
oral suspension is a white, cherry-flavored immediate-release
suspension containing 40 mg of posaconazole per mL and the following
inactive ingredients: polysorbate 80, simethicone, sodium benzoate,
sodium citrate dihydrate, citric acid monohydrate, glycerin, xanthan
gum, liquid glucose, titanium dioxide, artificial cherry flavor, and
purified water.
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
4-(4-(4-(4-(((3R,5R)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)oxolan-3-yl)methoxy)phenyl)piperazin-1-yl)phenyl)-2-((2S,3S)-2-hydroxypentan-3-yl)-1,2,4-triazol-3-one
| |
| Clinical data | |
| Trade names | Noxafil, Posanol |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a607036 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category | |
| Legal status |
|
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | High |
| Protein binding | 98 to 99% |
| Metabolism | Hepatic glucuronidation |
| Biological half-life | 16 to 31 hours |
| Excretion | Fecal (77%) and renal (14%) |
| Identifiers | |
| CAS Registry Number | 171228-49-2 |
| ATC code | J02AC04 |
| PubChem | CID: 147912 |
| DrugBank | DB01263 |
| ChemSpider | 130409 |
| UNII | 6TK1G07BHZ |
| KEGG | D02555 |
| ChEBI | CHEBI:64355 |
| ChEMBL | CHEMBL1397 |
| Synonyms | 4-{4-[4-(4-{[(5R)-5-(2,4-difluorophenyl)-5-(1H-1,2,4-triazol-1-ylmethyl)oxolan-3-yl]methoxy}phenyl)piperazin-1-yl]phenyl}-1-[(2S,3S)-2-hydroxypentan-3-yl]-4,5-dihydro-1H-1,2,4-triazol-5-one |
| Chemical data | |
| Formula | C37H42F2N8O4 |
| Molecular mass | 700.778 g/mol |

1H NMR PREDICT



13C NMR PREDICT


COSY PREDICT

| CN101824009A * | May 27, 2010 | Sep 8, 2010 | 北京德众万全药物技术开发有限公司 | Simple preparation method for posaconazole and piperazine intermediate thereof |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015011224A1 * | Jul 24, 2014 | Jan 29, 2015 | Sandoz Ag | Improved process for the preparation of crystalline form iv of posaconazole |
12

No comments:
Post a Comment