
1 Mirogabalin
2 Pregabalin
3 Atagabalin
4
5
WILL BE UPDATED
1
Mirogabalin


Mirogabalin, A-2000700, DS-5565
1138245-13-2, C12H19NO2, 209.28
[(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
2-[(1R,5S,6S)-6-(aminomethyl)-3-ethyl-6-bicyclo[3.2.0]hept-3-enyl]acetic acid
UNII-S7LK2KDM5U
| Originator |
Daiichi Sankyo
|
|---|---|
| Therapeutic Claim |
Treatment of fibromyalgia
|

| Class |
Analgesic drugs (small molecules)
|
|---|---|
| Mechanism of action |
CACNA2D1 protein modulators
|

- SEE
- [(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid benzenesulfonate

- [(1S,5R,6R)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid , optical isomer of the compound
 UNDESIRED
UNDESIRED
Mirogabalin (DS-5565) is a drug developed by Daiichi Sankyo and related to drugs such as gabapentin and pregabalin. Similarly to these drugs, mirogabalin binds to the α2δ calcium channels (1 and 2), but with significantly higher potency than pregabalin. It has shown promising results in Phase II clinical trials for the treatment of diabetic peripheral neuropathic pain,[1][2] and is currently in Phase III trials.
Mirogabalin, a voltage-dependent calcium channel subunit alpha-2/delta-1 ligand, is in phase III clinical trials at Daiichi Sankyo for the treatment of pain associated with fibromyalgia. The company is also conducting phase III clinical studies for the treatment of chronic pain and pain associated with diabetic peripheral neuropathy.

Mirogabalin, a voltage-dependent calcium channel subunit alpha-2/delta-1 ligand, is in phase III clinical trials at Daiichi Sankyo for the treatment of pain associated with fibromyalgia. The company is also conducting phase III clinical studies for the treatment of chronic pain and pain associated with diabetic peripheral neuropathy.
Mirogabalin besylate
cas 1138245-21-2
UNII: 01F4FRP8YL
C12-H19-N-O2.C6-H6-O3-S, 367.4635
Bicyclo(3.2.0)hept-3-ene-6-acetic acid, 6-(aminomethyl)-3-ethyl-, (1R,5S,6S)-, benzenesulfonate (1:1)
SEETert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate.....http://www.google.com/patents/US20140094623?cl=zh
PATENT
WO 2009041453https://www.google.co.in/patents/EP2192109A1
- (Example
21)
[(1S,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic
acid (exemplary compound No: 8, optically active form of the compound of
Example 8)

- Tert-butyl (±)-[(1R,5S,6S)-3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (230 g, 778 mmol) was resolved using Chiralpak IC (N-Hex:EtOH=98:2, 1.0 mL/min, 40°C) manufactured by Daicel Chemical Industries, Ltd. to respectively obtain 115 g of a peak 1 (retention time: 5.2 min) and 93.7 g of a peak 2 (retention time: 6.3 min).
- Tert-butyl [(1R,5S,6S)-3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (peak 1, 7.0 g, 23.7 mmol) was dissolved in ethanol (60 mL) and water (21 mL). To the solution, iron powder (13.27 g, 237 mmol) and ammonium chloride (628.1 mg, 11.9 mmol) were added, and the mixture was stirred for 5.5 hours under heating to reflux. The mixture was allowed to cool, then diluted with saturated saline, a saturated aqueous solution of sodium bicarbonate, and ethyl acetate, and filtered through Celite to remove insoluble matter. The filtrate was separated into organic and aqueous layers. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate. Then, the solvent was distilled off under reduced pressure to obtain a pale yellow oil substance (7.02 g). This substance was dissolved in dichloromethane (200 mL). To the solution, (Boc)2O (5.25 g, 25 mmol) and triethylamine (5.01 g, 50 mmol) were added, and the mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure, and the residue was then purified by silica gel chromatography to obtain the title compound of interest as a pale yellow oil substance (8.82 g, <100%). (21-c) [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
- A 4 N hydrochloric acid-ethyl acetate solution (100 mL) was added to tert-butyl (1R,5S,6S)-[6-(tert-butoxycarbonylaminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate (9.82 g, 23.7 mmol), and the mixture was stirred at room temperature for 1 hour. Then, the solvent was distilled off under reduced pressure. The residue was dissolved in dichloromethane. To the solution, triethylamine was added dropwise, and the resulting powder was collected by filtration, then washed with dichloromethane, and then dried to obtain 4.02 g of a white powder. This powder was washed with ethanol and ethyl acetate to obtain the title compound of interest as a white powder (2.14 g, 43%).
- (Example 31)
[(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic
acid benzenesulfonate (exemplary compound No: 8, optically active
benzenesulfonate)
- (1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid (4.50 g, 20.6 mmol) was dissolved by heating in a 1 M aqueous solution (22.7 mL) of benzenesulfonic acid monohydrate, and the solution was then allowed to cool to room temperature. The resulting solid was collected by filtration. The solid was washed with water (15 mL) and then dried using a vacuum pump to obtain the compound of interest as a colorless solid (6.45 g, 77%).
PATENT
JP 2010241796PATENT
WO 2012169475- Reference Example 1[6-Aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
- Sodium hydride (>63% oil, 2.09 g, 55 mmol) was added to a solution of ethyl 3-oxohexanoate (7.91 g, 50 mmol) in tetrahydrofuran (50 mL) under ice cooling, and the mixture was stirred in this state for 10 minutes. To the reaction solution, n-butyllithium (1.58 M solution in hexane, 34.8 mL, 55 mmol) was added dropwise, and the mixture was further stirred for 10 minutes under ice cooling. Then, allyl bromide (4.7 mL, 55 mmol) was added thereto, and the mixture was stirred in this state for 1 hour and then further stirred at room temperature for 4 hours. To the reaction solution, 1 N hydrochloric acid and a saturated aqueous solution of ammonium chloride were added, followed by extraction with n-pentane. The organic layer was washed with saturated saline and dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was dissolved in ethanol (80 mL). To the solution, sodium borohydride (1.51 g, 40 mmol) was added under ice cooling, and the mixture was stirred in this state for 2 hours. 1 N hydrochloric acid (50 mL) was added thereto, and the mixture was stirred for 30 minutes. Then, saturated saline was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography to obtain the compound of interest as a pale yellow oil substance (3.64 g, 37%, mixture of diastereomers).
- 1H-NMR (400 MHz, CDCl3): δ ppm: 0.91 (3H, t, J=7.5 Hz), 1.28 (3H, t, J=7.2 Hz), 1.43-1.55 (2H, m), 1.98-2.28 (2H, m), 2.45-2.48 (2H, m), 2.88-2.93 (1H, m), 4.07-4.10 (1H, m), 4.10-4.20 (2H, m), 5.01-5.09 (2H, m), 5.75-5.86 (1H, m).
- Ethyl 4-ethyl-3-hydroxyhept-6-enoate (3.64 g, 18.2 mmol) was dissolved in a 2 N solution of potassium hydroxide in methanol (120 mL), and the solution was stirred overnight at room temperature. From the reaction solution, the solvent was distilled off under reduced pressure. To the residue, a 1 N aqueous sodium hydroxide solution (200 mL) was then added, followed by extraction with diethyl ether. The aqueous layer was made acidic by the addition of concentrated hydrochloric acid under ice cooling, followed by extraction with diethyl ether again. The organic layer was washed with saturated saline and dried over anhydrous magnesium sulfate. Then, the solvent was distilled off under reduced pressure to obtain the compound of interest as a pale yellow oil substance (3.14 g, <100%, mixture of diastereomers).
- 1H-NMR (400 MHz, CDCl3): δ ppm: 0.91-0.96 (3H, m), 1.39-1.52 (3H, m), 2.01-2.28 (2H, m), 2.52-2.55 (2H, m), 4.05-4.15 (2H, m), 5.03-5.10 (2H, m), 5.74-5.86 (1H, m).
- 4-Ethyl-3-hydroxyhept-6-enoic acid (3.13 g, 18.2 mmol) was dissolved in acetic anhydride (15 mL). To the solution, potassium acetate (4.27 g, 43.6 mmol) was added, and the mixture was stirred at room temperature for 100 minutes. The reaction solution was heated to reflux and stirred for 3.5 hours to form “3-ethylbicyclo[3.2.0]hept-6-en-6-one” in the reaction solution. To the reaction solution, ice water and toluene were then added, and this mixture was stirred overnight at room temperature. The mixture was separated into aqueous and organic layers by the addition of saturated saline (50 mL) and toluene (20 mL). Then, the organic layer was washed with a 1 N aqueous sodium hydroxide solution and saturated saline in this order, then dried over anhydrous magnesium sulfate, and filtered. The filtrate was added to a reaction solution prepared by adding sodium hydride (>65% oil, 761.9 mg, 20 mmol) to a solution of tert-butyl dimethoxyphosphorylacetate (4.48 g, 20 mmol) in tetrahydrofuran (50 mL) under ice cooling, and the mixture was further stirred for 1 hour. The reaction solution was separated into aqueous and organic layers by the addition of a saturated aqueous solution of ammonium chloride and saturated saline. The aqueous layer was subjected to extraction with ethyl acetate. The organic layers were combined, then washed with saturated saline, and then dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography to obtain the compound of interest as a pale yellow oil substance (1.32 g, 31%, E/Z mixture).
- 1H-NMR (400 MHz, CDCl3): δ ppm:
- Major isomer: 1.06 (3H, t, J=7.4 Hz), 1.45 (9H, s), 2.07-2.22 (3H, m), 2.59-2.70 (2H, m), 2.87-2.96 (1H, m), 3.30 (1H, ddt, J=8.6, 18.4, 2.7 Hz), 3.86-3.88 (1H, m), 5.22-5.23 (1H, m), 5.45-5.47 (1H, m).
- Minor isomer: 1.08 (3H, t, J=7.3 Hz), 1.49 (9H, s), 2.07-2.21 (3H, m), 2.43-2.47 (1H, m), 2.59-2.70 (1H, m), 2.75-2.85 (1H, m), 2.87-2.96 (1H, m), 4.28-4.31 (1H, m), 5.35-5.38 (1H, m), 5.45-5.47 (1H, m).
- Tert-butyl [3-ethylbicyclo[3.2.0]hept-3-en-6-ylideneacetate (1.32 g, 5.63 mmol) was dissolved in nitromethane (7 mL). To the solution, 1,8-diazabicyclo[5.4.0]undec-7-ene (1.2 mL, 7.3 mmol) was added, and the mixture was heated with stirring at 50 to 60° C. for 7 hours. The mixture was allowed to cool, and a saturated aqueous solution of potassium dihydrogen phosphate was then added thereto, followed by extraction with ethyl acetate. Then, the organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography to obtain the compound of interest as a colorless oil substance (1.39 g, 84%).
- 1H-NMR (400 MHz, CDCl3): δ ppm: 1.09 (3H, t, J=7.4 Hz), 1.46 (9H, s), 1.52 (1H, dd, J=7.6, 13.2 Hz), 2.06 (1H,d, 16.6 Hz), 2.14 (2H, q, J=7.4 Hz), 2.30 (1H, ddd, J=2.4, 7.6, 13.2 Hz), 2.47 (2H, s), 2.49 (1H, dd, J=7.6,16.6 Hz), 2.86 (1H, quint, J=7.6 Hz), 3.21-3.22 (1H, m), 4.75 (1H, d, J=11.7 Hz), 4.84 (1H, d, J=11.7 Hz), 5.27 (1H, s).
- Tert-butyl [3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (1.09 g, 4.71 mmol) was dissolved in ethanol (10 mL) and water (5 mL). To the solution, iron powder (1.32 g, 23.5 mmol) and ammonium chloride (249.6 mg, 4.71 mmol) were added, and the mixture was stirred for 2 hours under heating to reflux. The mixture was allowed to cool, then diluted with saturated saline, a saturated aqueous solution of sodium bicarbonate, and ethyl acetate, and filtered through Celite to remove insoluble matter. The filtrate was separated into organic and aqueous layers. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate, and the solvent was then distilled off under reduced pressure. To the residue, a 4 N solution of hydrochloric acid in ethyl acetate (20 mL) was added, and the mixture was stirred at room temperature for 1 hour. Then, the solvent was distilled off under reduced pressure. The residue was suspended in dichloromethane. To the suspension, triethylamine was added dropwise, and the resulting powder was collected by filtration, then washed with dichloromethane, and then dried to obtain the compound of interest as a white powder (425.1 mg, 43%).
- 1H-NMR (400 MHz, CD3OD): δ ppm: 1.10 (3H, t, J=7.4 Hz), 1.48 (1H, dd, J=7.5, 12.5 Hz), 2.03-2.08 (2H, m), 2.14 (2H, q, J=7.4 Hz), 2.46 (1H, d, J=16.2 Hz), 2.46-2.53 (1H, m), 2.51 (1H, d, J=16.2 Hz), 2.85 (1H, quint, J=7.5 Hz), 3.09-3.10 (1H, m), 3.14 (1H, d, J=13.0 Hz), 3.18 (1H, d, J=13.0 Hz), 5.38 (1H, dd, J=1.7, 3.7 Hz).
- (Step of Performing Optical Resolution from Diastereomeric Mixture)
- Reference Example 2Tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate
- Acetonitrile (4.7 L, 8.6 v/w) was added to tert-butyl [6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate (627.0 g, net: 543.6 g, 2.05 mol, 85:15 diastereomeric mixture), and the mixture was stirred at 40° C. To the reaction solution, D-mandelic acid (116.3 g, 0.76 mmol, 0.37 eq.) was added, and the mixture was stirred at 40° C. for 1 hour and then allowed to cool slowly to 3° C. After stirring at 3° C. for 1 hour, the resulting crystal was collected by filtration. Then, the crystal was dried under reduced pressure under the condition of 40° C. to obtain tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate as a white powder (251.2 g, yield: 29.4%, 97.6% ee, 99.6% de).
- 1H-NMR (400 MHz, DMSO-d6) δ ppm: 1.04 (3H, t, J=7.6 Hz), 1.28-1.35 (1H, m), 1.39 (9H, s), 1.96-2.11 (4H, m), 2.28 (1H, d, J=15.6 Hz), 2.33 (1H, d, J=15.6 Hz), 2.36-2.40 (1H, m), 2.72 (1H, quint, J=7.6 Hz), 3.00 (1H, d, J=13.2 Hz), 3.03 (1H, d, J=13.2 Hz), 3.31 (1H, br s), 4.54 (1H, s), 5.21-5.23 (1H, m), 7.13-7.25 (3H, m), 7.35-7.37 (2H, m).
- [α]20 D −104.4° (C=0.108, MeOH).
- Anal. calcd for C24H35NO5: C, 69.04; H, 8.45; N, 3.35; Found C, 69.15; H, 8.46; N, 3.46.



PATENT
WO 2012169474PATENT
WO2015005298https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015005298&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=FullText
[Step D-2]
a compound having the formula (Va) (and its enantiomers), and to carry out optical resolution by chloride with optically active organic amine, and is a process for preparing a compound having the general formula (VIa) .
[Formula 19] The solvent used in this step, MTBE, CPME, ethers such as THF; aromatic hydrocarbons such as toluene; esters such as ethyl acetate; EtOH, alcohols such as diisopropyl alcohol CH; s three nitriles such as CN; ketones such as acetone; or is a mixed solvent of these solvents and water, preferably toluene, ethyl acetate, CH 3 CN, are MTBE, More preferably, toluene, MTBE. Optically active organic amine used in this step, preferably, (1R, 2R) -trans-1- amino-2-indanol, (S) -2- phenylglycinol, (R) -1- ( p- tolyl) ethylamine, (1R, 2S) -2- amino-1,2-diphenyl ethanol, (S) -1- (2- naphthyl) ethylamine, (R) -1- (4- bromophenyl) ethylamine, (1S, 2R) - (+) - 1- amino-2-indanol is a L- phenylalaninol, etc., more preferably, (1R, 2R) -trans-1- amino-2-indanol, (S ) -2-phenylglycinol. Equivalent of the optically active organic amine to be used have the general formula (Va) compound having a relative (and its enantiomers) are 0.5-1.1 equivalents. The reaction temperature of this step is such as about 0-50 ℃, preferably, after aging the crystals at about 10-30 ℃, is obtained by filtering the compound of formula (VIa). The time required to chloride present step is not particularly limited, but is usually 4 to about 48 hours. In this step, (1) with respect to (Va) compound with (and its enantiomers), directly to a compound of formula (VIa) with the desired configuration by the action of the above-mentioned optically active amine How to get, or, with respect to (2) compounds having formula (Va) (or its enantiomer), first, quinine, (1S, 2S) -trans-1- amino-2-indanol, (R) -2- by the action of an optically active amine such as phenylglycinol, it allowed to temporarily deposit the enantiomer having the unnecessary configuration, after removing the precipitate by filtration, against followed by compound obtained from the filtrate, (1R, 2R ) -trans-1- amino-2-indanol, by the action of optically active amines such as (S) -2- phenylglycinol, to precipitate the salt of the compound of formula (VIa) with the desired configuration How to get Te, one of the methods is used.
 Known
compounds having the general formula (Va) which are used in the above
Step D-1 or step D-2, which can be prepared according to step A-C, as
otherwise, it is disclosed in Patent Document 5 It can be prepared by
method (the following scheme).
Known
compounds having the general formula (Va) which are used in the above
Step D-1 or step D-2, which can be prepared according to step A-C, as
otherwise, it is disclosed in Patent Document 5 It can be prepared by
method (the following scheme).
[Formula 20] specific production method according to the present method will be described later as a reference example.

a compound having the formula (Va) (and its enantiomers), and to carry out optical resolution by chloride with optically active organic amine, and is a process for preparing a compound having the general formula (VIa) .
[Formula 19] The solvent used in this step, MTBE, CPME, ethers such as THF; aromatic hydrocarbons such as toluene; esters such as ethyl acetate; EtOH, alcohols such as diisopropyl alcohol CH; s three nitriles such as CN; ketones such as acetone; or is a mixed solvent of these solvents and water, preferably toluene, ethyl acetate, CH 3 CN, are MTBE, More preferably, toluene, MTBE. Optically active organic amine used in this step, preferably, (1R, 2R) -trans-1- amino-2-indanol, (S) -2- phenylglycinol, (R) -1- ( p- tolyl) ethylamine, (1R, 2S) -2- amino-1,2-diphenyl ethanol, (S) -1- (2- naphthyl) ethylamine, (R) -1- (4- bromophenyl) ethylamine, (1S, 2R) - (+) - 1- amino-2-indanol is a L- phenylalaninol, etc., more preferably, (1R, 2R) -trans-1- amino-2-indanol, (S ) -2-phenylglycinol. Equivalent of the optically active organic amine to be used have the general formula (Va) compound having a relative (and its enantiomers) are 0.5-1.1 equivalents. The reaction temperature of this step is such as about 0-50 ℃, preferably, after aging the crystals at about 10-30 ℃, is obtained by filtering the compound of formula (VIa). The time required to chloride present step is not particularly limited, but is usually 4 to about 48 hours. In this step, (1) with respect to (Va) compound with (and its enantiomers), directly to a compound of formula (VIa) with the desired configuration by the action of the above-mentioned optically active amine How to get, or, with respect to (2) compounds having formula (Va) (or its enantiomer), first, quinine, (1S, 2S) -trans-1- amino-2-indanol, (R) -2- by the action of an optically active amine such as phenylglycinol, it allowed to temporarily deposit the enantiomer having the unnecessary configuration, after removing the precipitate by filtration, against followed by compound obtained from the filtrate, (1R, 2R ) -trans-1- amino-2-indanol, by the action of optically active amines such as (S) -2- phenylglycinol, to precipitate the salt of the compound of formula (VIa) with the desired configuration How to get Te, one of the methods is used.
Known
compounds having the general formula (Va) which are used in the above
Step D-1 or step D-2, which can be prepared according to step A-C, as
otherwise, it is disclosed in Patent Document 5 It can be prepared by
method (the following scheme).[Formula 20] specific production method according to the present method will be described later as a reference example.
[Step E]
Formula (V) or a compound having the general formula (VI) from (and / or its enantiomer) is a process for preparing a compound of formula (VII) (and / or its enantiomer), the general formula (V) is a compound having (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst, reduction with a solvent, or a compound having the general formula (VI) (and / or its enantiomer) solution compounds having the general formula (V) obtained by salt (and / or its enantiomer) solution, under a hydrogen atmosphere to carry out a reduction reaction in the presence of a metal catalyst, by a compound of formula (VII) This is a method of manufacturing a.
Formula 21] (1) Kaishio step formula compound with a (VI) (and / or its enantiomer) is suspended in an organic solvent, washed with an aqueous solution obtained by adding an acid, by liquid separation and the organic layer , compounds having general formula (V) (and / or its enantiomer) solution containing it can get. The solvent used in this step include aromatic hydrocarbons such as toluene, ethers such as MTBE, an ester such as ethyl acetate, and the like, preferably toluene, or is MTBE. Acid used in this step is not particularly limited, hydrochloric acid, sulfuric acid, phosphoric acid, citric acid, malonic acid can be used.

Formula (V) or a compound having the general formula (VI) from (and / or its enantiomer) is a process for preparing a compound of formula (VII) (and / or its enantiomer), the general formula (V) is a compound having (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst, reduction with a solvent, or a compound having the general formula (VI) (and / or its enantiomer) solution compounds having the general formula (V) obtained by salt (and / or its enantiomer) solution, under a hydrogen atmosphere to carry out a reduction reaction in the presence of a metal catalyst, by a compound of formula (VII) This is a method of manufacturing a.
Formula 21] (1) Kaishio step formula compound with a (VI) (and / or its enantiomer) is suspended in an organic solvent, washed with an aqueous solution obtained by adding an acid, by liquid separation and the organic layer , compounds having general formula (V) (and / or its enantiomer) solution containing it can get. The solvent used in this step include aromatic hydrocarbons such as toluene, ethers such as MTBE, an ester such as ethyl acetate, and the like, preferably toluene, or is MTBE. Acid used in this step is not particularly limited, hydrochloric acid, sulfuric acid, phosphoric acid, citric acid, malonic acid can be used.
(2) the reduction reaction step
compounds having the general formula (V) (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst was reduced in a solvent, a cyano group (or a nitro group) and an amino group It is converted into, and is a step for preparing a compound of formula (VII). This reaction is usually carried out in a neutral or basic conditions.
The solvent used in this step include aromatic hydrocarbons such as toluene, MTBE, ethers such as THF, alcohols of C1-C4, or is water, preferably toluene, MTBE, or water , and the Particularly preferred is water.
Metal catalyst used in this step, vinegar Sanskrit nickel, sponge cobalt, or palladium - is carbon, preferably, sponge nickel (eg, Kawaken Fine Chemicals Co., Ltd. of PL-9T, NDT-65, NDT- 90, NDHT-90M, NDHT-M3, and the like, or, Nikko Rika Co., Ltd. R-100, R-200, such as R-205, R-211, R-2311), or, sponge cobalt (for example, the river Research ODHT-60 manufactured by Fine Chemical Co., Ltd., OFT-55, or the like, or is a Nikko Rika Co., Ltd. R-400, R-400K, such as R-401, R-455, such as A-8B46 manufactured by Johnson Matthey) .
In this step, when carrying water as a solvent is usually added to the base. As the base used, preferably an inorganic base, particularly preferred are lithium hydroxide, sodium hydroxide, alkali metal hydroxides such as potassium hydroxide.
In this step, by the addition of aqueous ammonia, it is possible to improve the yield, it is not necessarily added aqueous ammonia.
In this step, by the addition of dimethyl polysiloxane, it is possible to suppress the generation of bubbles from the reaction liquid, it is not necessarily added dimethylpolysiloxane.
The reaction temperature in this step is about 20-60 ℃, preferably, is about 30-50 ℃.
The reaction time of this step, the raw material is not particularly limited as long as it is a time that is substantially consumed, it is usually 2 to about 12 hours.
In this step, after the completion of the reaction, the catalyst was removed by filtration, by adding an acid to the filtrate, by then crystallizing the compound of formula (VII), and filtering and washing the precipitate, pure products a you can get.
compounds having the general formula (V) (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst was reduced in a solvent, a cyano group (or a nitro group) and an amino group It is converted into, and is a step for preparing a compound of formula (VII). This reaction is usually carried out in a neutral or basic conditions.
The solvent used in this step include aromatic hydrocarbons such as toluene, MTBE, ethers such as THF, alcohols of C1-C4, or is water, preferably toluene, MTBE, or water , and the Particularly preferred is water.
Metal catalyst used in this step, vinegar Sanskrit nickel, sponge cobalt, or palladium - is carbon, preferably, sponge nickel (eg, Kawaken Fine Chemicals Co., Ltd. of PL-9T, NDT-65, NDT- 90, NDHT-90M, NDHT-M3, and the like, or, Nikko Rika Co., Ltd. R-100, R-200, such as R-205, R-211, R-2311), or, sponge cobalt (for example, the river Research ODHT-60 manufactured by Fine Chemical Co., Ltd., OFT-55, or the like, or is a Nikko Rika Co., Ltd. R-400, R-400K, such as R-401, R-455, such as A-8B46 manufactured by Johnson Matthey) .
In this step, when carrying water as a solvent is usually added to the base. As the base used, preferably an inorganic base, particularly preferred are lithium hydroxide, sodium hydroxide, alkali metal hydroxides such as potassium hydroxide.
In this step, by the addition of aqueous ammonia, it is possible to improve the yield, it is not necessarily added aqueous ammonia.
In this step, by the addition of dimethyl polysiloxane, it is possible to suppress the generation of bubbles from the reaction liquid, it is not necessarily added dimethylpolysiloxane.
The reaction temperature in this step is about 20-60 ℃, preferably, is about 30-50 ℃.
The reaction time of this step, the raw material is not particularly limited as long as it is a time that is substantially consumed, it is usually 2 to about 12 hours.
In this step, after the completion of the reaction, the catalyst was removed by filtration, by adding an acid to the filtrate, by then crystallizing the compound of formula (VII), and filtering and washing the precipitate, pure products a you can get.
[Step F]
compounds
having the formula (VII) (and / or its enantiomer), to produce the
presence of an organic acid and a solvent, a compound having formula
(VIII) is allowed to form salts with (and / or its enantiomer) It is a
method.
Chemical Formula 22] The solvent used in this step include water, anisole, aqueous acetone, water CH 3 CN, MTBE water - acetone, anisole - acetate, anisole - acetone, anisole - acetate - acetone, acetone - water -CH 3 CN single like, or it is a mixed solvent, preferably, water, anisole. The organic acid used in this step is an organic acid that is pharmacologically today preferably a benzenesulfonic acid. Equivalent of the organic acid used in this step is preferably a compound having the formula (VII) with respect to (and / or its enantiomer) is about 1.00-1.10 equivalents. This step is carried out in the range of usually about -15-50 ℃, preferably, after aging the crystals at a temperature of about -10-30 ℃, filtration, by washing, the general formula (VIII) a compound having a (and / or its enantiomers) get. The time required for chloride in this step is not particularly limited, but is usually 1 to about 24 hours.
 In
the present invention, compounds having formula (IX) prepared via the
process F from Step A (and / or its enantiomer) may be very produced as
pure compounds. Compounds of formula (IX) which can be obtained by the
present invention typically have a quality below.
In
the present invention, compounds having formula (IX) prepared via the
process F from Step A (and / or its enantiomer) may be very produced as
pure compounds. Compounds of formula (IX) which can be obtained by the
present invention typically have a quality below.
Chemical Formula 22] The solvent used in this step include water, anisole, aqueous acetone, water CH 3 CN, MTBE water - acetone, anisole - acetate, anisole - acetone, anisole - acetate - acetone, acetone - water -CH 3 CN single like, or it is a mixed solvent, preferably, water, anisole. The organic acid used in this step is an organic acid that is pharmacologically today preferably a benzenesulfonic acid. Equivalent of the organic acid used in this step is preferably a compound having the formula (VII) with respect to (and / or its enantiomer) is about 1.00-1.10 equivalents. This step is carried out in the range of usually about -15-50 ℃, preferably, after aging the crystals at a temperature of about -10-30 ℃, filtration, by washing, the general formula (VIII) a compound having a (and / or its enantiomers) get. The time required for chloride in this step is not particularly limited, but is usually 1 to about 24 hours.
In
the present invention, compounds having formula (IX) prepared via the
process F from Step A (and / or its enantiomer) may be very produced as
pure compounds. Compounds of formula (IX) which can be obtained by the
present invention typically have a quality below.
The content of the diastereomer represented by the formula (X): 0.1% less than the
content of the enantiomers represented by the formula (XI): 1.0% less than
the formula (XII) and the double bond represented by the formula (XIII) The total content of regioisomers: less than 0.5%
(Note that each content is calculated from the area percentage of the free form of formula (IX) (VII) in the by test High Performance Liquid Chromatography)
[formula 23] [of 24]


content of the enantiomers represented by the formula (XI): 1.0% less than
the formula (XII) and the double bond represented by the formula (XIII) The total content of regioisomers: less than 0.5%
(Note that each content is calculated from the area percentage of the free form of formula (IX) (VII) in the by test High Performance Liquid Chromatography)
[formula 23] [of 24]
Next,
the present invention is described by examples in detail, the present
invention is, which however shall not be construed as limited thereto.
The internal standard substance in a magnetic resonance spectra (NMR), and using tetramethylsilane and abbreviations indicate the multiplicity, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and brs = It shows a broad singlet.
In the name of the compound, "R" and "S" indicate the absolute configuration at the asymmetric carbon. Furthermore, "RS" and "SR" indicates that the asymmetric carbon atom is racemic. In addition, "(1RS, and 5SR) -" if such a can shows the relative arrangement of the 1-position and the 5-position, as well shows only one of the diastereomers, its diastereomers are racemic We show that.
In the name of the compound, "E" and "Z" indicates the arrangement of positional isomers in the structure of the compound having a position isomerism.
The internal standard substance in a magnetic resonance spectra (NMR), and using tetramethylsilane and abbreviations indicate the multiplicity, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and brs = It shows a broad singlet.
In the name of the compound, "R" and "S" indicate the absolute configuration at the asymmetric carbon. Furthermore, "RS" and "SR" indicates that the asymmetric carbon atom is racemic. In addition, "(1RS, and 5SR) -" if such a can shows the relative arrangement of the 1-position and the 5-position, as well shows only one of the diastereomers, its diastereomers are racemic We show that.
In the name of the compound, "E" and "Z" indicates the arrangement of positional isomers in the structure of the compound having a position isomerism.
"EZ" and "ZE"
indicates that it is a mixture of regioisomers. Way more notation, is in
accordance with the conventions in this area of the normal.
(Example 1)
(2EZ)-3-ethoxy -2 - [(1R, 5S) -3- Echirubishikuro [3.2.0] hept-3-en-6-ylidene] -3-oxo-propanoic acid (2EZ) -3-Ethoxy-2 - [(1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] -3-Oxopropanoic acid [of 25] malonic acid mono ethyl ester (2.9 g, AlCl in THF (20 mL) solution of 22.0 mmol) 3
(3.9 g, after addition of 29.4 mmol) in -10 ° C, (1R, 5S)
-3-Ethylbicyclo [3.2.0] hept-3-en- 6-one (2.0 g, 14.7 mmol) was added
and stirred for 25 h at -10 ° C. Under ice-cooling After stirring was
added with water (10 mL) CPME and (10 mL), and the organic layer was
separated and aqueous layer 1 1 25 ° C. The aqueous layer 1 was
extracted with CPME (20 mL), the organic layer 2 was separated and the
organic layer was combined with the organic layer one. After washing the
combined organic layers with 1 N hydrochloric acid (6 mL), and
concentrated under reduced pressure at an external temperature of 40 °
C, to give the title compound (4.8 g) as a crude product. 1 H NMR (CDCl 3
) (400 MHz): delta = 1.07 (3H, t, J = 7.6 Hz), 1.35 (1.5H, t, 7.2 Hz),
1.41 (1.5H, t, 7.2 Hz), 2.08- 2.16 (2H, m), 2.23-2.31 (1H, m), 2.67-2.75
(1H, m), 2.83-3.05 (2H, m), 3.40-3.48 (0.5H, m), 3.57-3.64 (0.5H , m),
4.27-4.41 (3H, m), 5.29 (0.5H, s), 5.50 (0.5H, s)

(Example 2) [(RS, 5SR)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] -3-oxo propanedioic acid dimethyl (racemic) Dimethyl [(RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] Propanedioate (Racemate) [of 26] THF for (3.2 mL), TiCl at 0 ° C 4
(0.175 mL, 1.60 mmol) a It was then added and stirred for 20 minutes.
Subsequently (1RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (112 mg,
0.819 mmol), dimethyl malonate (113 μL, 0.989 mmol) was added and
stirred for 50 min After, it was added pyridine (265 μL, 3.28 mmol).
After 1 hour stirring at 0 ° C, and subjected to stirring overnight with
warming to room temperature, quenched with water (6 mL), and extracted
three times with toluene (6 mL). The toluene layer saturated aqueous
sodium bicarbonate solution (6 mL), washed with saturated brine (6 mL),
after distilling off the solvent, PTLC (hexane: ethyl acetate = 5: 1)
and subjected to purification, the title compound as a colorless oil The
resulting (135 mg, 65%). 1 H NMR (CDCl 3 ) (400
MHz): delta = 1.05 (3H, D, J = 7.6 Hz), 2.09 (2H, Q, J = 7.6 Hz), 2.21
(1H, dd, J = 16.8, 3.2 Hz ), 2.60-2.76 (2H, m), 2.91 (1H, quint, J = 7.2
Hz), 3.30 (1H, ddd, J = 19.1, 8.4, 3.6 Hz), 3.73 (3H, s), 3.78 (3H, .
s), 4.29 (1H, M), 5.34 (1H, s) 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.2, 24.2, 32.6, 39.8, 42.7, 51.6, 51.7, 117.5, 120.9, 148.9 , 164.6, 164.9, 177.6.

(Example 7) [(1R, 5S)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] propane two acid diethyl Diethyl [(1R, 5S) -3-ethylbicyclo [3.2.0 ] hept-3-en-6-Ylidene] Propanedioate [of 31] to CPME (159 mL), 0 ° C with Ti (Oi-Pr) 4 (16.0 mL, 54.6 mmol) After addition of, TiCl 4
and stirred for 1 hour at (18.0 mL, 164 mmol) and over 8 minutes was
added dropwise 0 ° C. Then diethyl malonate (25.72 g, 161 mmol), was
added (1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (19.87 g, 146
mmol), 30-40 ° it was stirred for 4 hours at C. The reaction was
quenched with water (100 mL), and extracted with toluene (40 mL). After
the organic layer is concentrated under reduced pressure, to obtain a
crude product of the title compound as a yellow oil (43.61 g).

(Example 8) [(RS, 5SR)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] propane diacid di -tert- butyl (racemic) Di-tert-butyl [( RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] Propanedioate (Racemate) [of 32] with respect to THF (30 mL), and TiCl at 0 ° C 4
and (1.6 mL, and the mixture was stirred for 30 minutes was added 14.7
mmol). Subsequently (1RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one
(1.00 g, 7.34 mmol), malonic acid di -tert- butyl (1.91 g, 8.81 mmol)
was added After stirring for 1.5 hours, it was added pyridine (2.2 mL,
29.4 mmol). 0 ° 3.5 hours after stirring at C, and subjected to stirring
overnight with warming to room temperature, quenched with water (10
mL), and extracted two times with toluene (10 mL). After washed with
saturated brine (10 mL), the solvent was distilled off under reduced
pressure, silica gel column chromatography (hexane: ethyl acetate = 20:
1) and subjected to purification to give the title compound (2.26 g, 92%
). 1 H NMR (CDCl 3 ) (500 MHz): delta = 1.07 (3H,
t, J = 7.5 Hz), 1.47 (9H, s), 1.52 (9H, s), 2.06-2.14 (2H, M), 2.16
-2.24 (1H, m), 2.60-2.69 (2H, m), 2.90 (1H, quint, J = 7.0 Hz), 3.25
(1H, ddd, J = 18.6, 8.5, 3.5 Hz), 4.12-4.23 (1H , m), 5.36 (1H, s).

(Example 9) 5 - [(RS, 5SR)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] -2,2-dimethyl-1,3-dioxane -4-6- dione (racemic) 5 - [(RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] 2,2-dimethyl-1,3-dioxane-4-6-dione (Racemate) [of 33] THF for (80 mL), TiCl at 0 ° C 4
was stirred for 10 minutes was added (4.5 mL, 41 mmol). Subsequently
(1RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (2.81 g, 20.6 mmol),
Meldrum's acid (3.57 g, 24.8 mmol) was added and after stirring for 50
minutes , pyridine (6.53 g, 82.6 mmol) it was added. After 1.5 h
stirring at 0 ° C, and subjected to stirring overnight with warming to
room temperature, quenched with water (80 mL), and extracted three times
with toluene (50 mL). The organic layers with saturated brine (50 mL),
washed with 1 M HCl (10 mL), after distilling off the solvent, silica
gel column chromatography (hexane: ethyl acetate = 9: 1-6: 1) to perform
purification, as a white solid to give the title compound (4.51 g,
83.2%). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.05
(3H, t, J = 7.6 Hz), 1.69 (3H, s), 1.71 (3H, s), 2.11 (2H, Q, J = 7.6 Hz
), 2.20-2.35 (1H, m), 2.65-2.85 (1H, m), 2.92-3.13 (2H, m), 3.47-3.63
(1H, m), 4.45-4.59 (1H, m), 5.43 (1H , s). 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.1, 24.3, 27.59, 27.64, 34.1, 42.3, 42.8, 60.7, 104.4, 108.5, 119.4, 150.3, 160.1, 160.7.

(Example 10) [(1R, 5S, 6R)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane two acid dimethyl Dimethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate
[of 34] Dimethyl [(1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en-
6-ylidene] propanedioate (517 mg, 1.66 mmol) was dissolved in MeOH (5.2
mL), was added sodium cyanide (90 mg, 1.84 mmol) at room temperature and
stirred for 2 hours at room temperature. After quenching with 10%
aqueous acetic acid (5 mL), and extracted three times with ethyl acetate
(5 mL), the solvent was distilled off under reduced pressure to give
the title compound as an oil (667 mg). 1 H NMR (CDCl 3
) (400 MHz): delta = 1.08 (3H, t, J = 7.6 Hz), 1.80 (1H, dd, J = 12.4,
8.0 Hz), 2.01-2.22 (3H, M), 2.54 (1H, dd, J = 16.8, 7.6 Hz), 2.73 (1H,
ddd, J = 12.8, 8.8, 2.8 Hz), 3.18 (1H, quint, J = 7.6 Hz), 3.67 (1H, s),
3.78 ( . 3H, s), 3.82 (3H, s), 5.16-5.28 (1H, M) 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.2, 24.4, 32.1, 37.5, 39.2, 42.5, 52.9, 53.0 , 54.6, 55.0, 118.8, 123.2, 153.9, 166.62, 166.63.

(Example 11) [(1R, 5S, 6R)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane two acid diethyl Diethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate
[of 35] Diethyl obtained by the method shown in Example 7 [(1R, 5S)
-3-ethylbicyclo [3.2 .0] hept-3-en-6-Ylidene] Propanedioate crude
product (43.61 g, 146 mmol) was dissolved in EtOH (262 mL) and was added
sodium cyanide (7.15 g, 146 mmol) at room temperature , it was stirred
for 4 hours at room temperature. Acetate (8.76 g), after the reaction
quenched with water (180 mL), the solvent it was concentrated to
approximately 340 mL under reduced pressure. Water was added (80 mL),
then extracted three times with ethyl acetate (150 mL), the solvent was
distilled off under reduced pressure to give the title compound as an
oil (HPLC quantitative value: 44.29 g, 96.3% (( 1R, 5S) -3-Ethylbicyclo
[3.2.0] total yield from hept-3-en-6-one)). 1 H NMR (CDCl 3
) (400 MHz): delta = 1.07 (3H, t, J = 7.6 Hz), 1.28 (3H, t, J = 7.2
Hz), 1.31 (3H, t, J = 7.2 Hz), 1.80 (1H, dd, J = 12.6, 7.6 Hz),
2.01-2.19 (3H, m), 2.53 (1H, dd, J = 16.8, 7.6 Hz), 2.72 (1H, ddd, J =
12.6, 9.2, 2.8 Hz), 3.16 (1H, quint, J = 7.6 Hz), 3.61 (1H, s),
3.67-3.82 (1H, M), 4.15-4.33 (4H, M), 5.21-5.26 (1H, M). 13 C NMR (CDCl 3
) (100 MHz):. delta = 12.2, 14.0, 24.4, 32.2, 37.7, 39.3, 42.5, 55.0,
55.2, 62.00, 62.02, 119.0, 123.3, 153.7, 166.21, 166.23 (HPLC analysis
conditions) Diethyl [(1R, 5S, 6R) -6-cyano-3-ethylbicyclo [3.2.0]
hept-3-en-6-yl] propanedioate quantification method column: Cadenza
CW-C18 (Imtakt, 3 μm, 4.6 mm × 150 mm), 40 ° Cdetection wavelength: UV
205 nm mobile phase: MeCN: 0.1% AcOH aqueous solution = 10: 90-80: 20
(gradient) (0-2 min: MeCN 10%, 2-17 min: MeCN 10 → 80%, 17-25 min: MeCN
80%, 25-30 min: MeCN 80 → 10%, 40 min: STOP) measurement time: 40 min
flow rate: 1.0 mL / min retention time: Diethyl [(1R, 5S, 6R)
-6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate: 18.6 min,
Diethyl [(1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3 en-6-ylidene]
propanedioate: 19.7 min

(Example 12) [(1R, 5S, 6R)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane two acid diethyl Diethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate [of 36] under a nitrogen atmosphere, Ti (Oi-Pr) 4 (25.1 g, 88.11 mmol) the CPME (210 In addition to mL), TiCl it over 1 hour at 10-30 ° C 4
was added dropwise (29.0 mL, 264 mmol). After stirring for 30 minutes
at 25-30 ° C, was added diethyl malonate (38.8 g, 242 mmol) at 3-4 ° C,
stirred for 30 minutes at 1-4 ° C, (1R, 5S) -3-Ethylbicyclo- [3.2.0] In
addition hept-3-en-6-one a (30.0 g, 220 mmol) at 1-4 ° C, after which
the mixture was stirred for 2.5 hours at 32-33 ° C, ice cold cold water
(150 mL) was added thereto at the bottom, and the aqueous layer was
removed at room temperature. After washing with the organic layer 1 N
hydrochloric acid (60 mL), and concentrated under reduced pressure at an
external temperature of 40-45 ° C up to 120 mL, Diethyl [(1R, 5S)
-3-ethylbicyclo [3.2.0] hept- 3-en-6-ylidene] got CPME solution of
propanedioate. Under a nitrogen atmosphere, after addition of EtOH (150
mL) to the above solution was added sodium cyanide (10.8 g, 220 mmol),
and stirred for 4.5 h at 27-29 ° C. After cooling to 14 ℃, was added a
solution prepared by diluting concentrated sulfuric acid (10.8 g) in
water (60 mL), was added additional water and (150 mL). And the external
temperature 35-45 ° C under reduced pressure concentrated to 240 mL,
after removing the aqueous layer was added CPME (60 mL), the organic
layer was washed with 20% brine (60 mL), CPME of the title compound
solution was obtained (91.4%, HPLC quantitative value).

(Example 13) [(RS, 5SR, 6RS)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane diacid di -tert- butyl (racemic) Di tert-butyl [(RS, 5SR, 6RS) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate (Racemate)
[of 37] Di-tert-butyl [( 1RS, 5SR) -3-ethylbicyclo [3.2.0]
hept-3-en-6-ylidene] propanedioate (5.00 g, 14.9 mmol) was dissolved in
DMAc (50 mL), and sodium cyanide at room temperature (586 mg , it was
added 12.0 mmol), and stirred for 1 hour at room temperature. After
quenching with 1 M HCl (30 mL), and extracted three times with ethyl
acetate (50 mL), and the solvent was evaporated under reduced pressure.
Silica gel column chromatography (hexane: ethyl acetate = 20: 1) to give
to give the title compound as an oil (5.10 g, 94%). 1 H NMR (CDCl 3
) (400 MHz): delta = 1.06 (3H, t, J = 7.5 Hz), 1.46 (9H, s), 1.50 (9H,
s), 1.78 (1H, dd, J = 12.3, 8.0 Hz), 2.00-2.18 (3H, m), 2.51 (1H, dd, J =
17.0, 7.5 Hz), 2.68 (1H, ddd, J = 12.6, 8.5, 3.0 Hz), 3.13 (1H, quint, J
= 7.5 Hz), 3.40 (1H, s), 3.65-3.73 (1H, m), 5.24 (1H, s).

(Example 14) (RS, 5SR, 6RS)-6-(2,2-dimethyl-4,6-dioxo-1,3-dioxane-5-yl) -3-Echirubishikuro [3.2.0] hept - 3-en-6-carbonitrile (racemic) (RS, 5SR, 6RS)-6-(2,2-Dimethyl-4, 6-Dioxo-1,3-Dioxan-5-YL) -3-Ethylbicyclo [ 3.2.0] hept-3-ene-6-carbonitrile (Racemate)
[of 38] 5 - [(RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene]
-2, 2-dimethyl-1,3-dioxane-4,6-dione (100.8 mg, 0.384 mmol) was
dissolved in EtOH (1.0 mL) and was added sodium cyanide (22.0 mg, 0.449
mmol) at room temperature, room temperature in it was stirred for 3
hours. After quenching with phosphate buffer (pH 7) (5 mL), and
extracted three times with ethyl acetate (5 mL), the solvent was
distilled off under reduced pressure, a white solid to give the title
compound (23.6 mg, 21.2 %). 1 H NMR (CD 3 OD) (400
MHz): delta = 1.03 (3H, t, J = 7.6 Hz), 1.61 (3H, s), 1.92-2.25 (4H,
M), 2.45 (1H, dd, J = 16.8, 7.2 Hz), 2.66-2.80 (1H, m), 3.00 (1H, quint,
J = 7.6 Hz), 3.72-3.87 (1H, m), 4.85 (1H, s), 5.23-5.33 (1H, . M) 13 C NMR (CD 3 OD) (100 MHz): delta = 12.66, 12.69, 25.3, 34.1, 38.8, 39.4, 43.3, 57.0, 75.8, 102.9, 123.67, 123.70, 127.9, 150.5, 167.9.

(Example 15) [(RS, 5SR, 6SR)-3-ethyl-6- (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] ethyl acetate (racemic) Ethyl [(RS, 5SR, 6SR) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] acetate (Racemate)
[of 39] Diethyl [(RS, 5SR) -3-Ethylbicyclo [ 3.2.0]
hept-3-en-6-ylidene] propanedioate (256.0 mg, 0.920 mmol) was dissolved
in toluene (2.5 mL), was added DBU (152 mL), nitromethane (55 mL), at
room temperature for 17 time it was stirred. After quenching with 1 M
HCl (5 mL), and extracted three times with ethyl acetate (5 mL), and the
resulting ethyl acetate solution was washed with saturated brine (5
mL). The solvent was evaporated under reduced pressure, as a pale yellow
oily substance Diethyl [(1RS, 5SR, 6SR) -3-ethyl-6- (nitromethyl)
bicyclo- [3.2.0] hept-3-en-6-yl] propanedioate was obtained (336.9 mg).
The resulting Diethyl [(RS, 5SR, 6SR) -3-ethyl-6 (Nitromethyl) bicyclo
[3.2.0] hept-3-en-6-YL] - Propanedioate a (336.9 mg) DMSO and (3.4 mL)
It was dissolved in water (50 μL, 2.78 mmol), sodium chloride (64.8 mg,
1.11 mmol) was added, followed by 10 hours heated and stirred at 140 °
C. After cooling to room temperature, the reaction was quenched with 1 M
HCl (5 mL), was extracted three times with ethyl acetate (5 mL), and
the resulting ethyl acetate solution was washed with saturated brine (5
mL). The solvent was evaporated under reduced pressure to give the title
compound as a brown oily substance (261.6 mg, 2 process overall yield
72.4%). Diethyl [(RS, 5SR, 6SR) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0]
hept-3-en-6-YL] Propanedioate 1 H NMR (CDCl 3 )
(400 MHz): delta = 1.08 (3H, t, J = 7.6 Hz), 1.17-1.35 (6H, m), 1.73
(1H, dd, J = 13.2, 7.6 Hz), 2.05 (1H, d, J = 16.4 Hz), 2.05-2.22 (2H,
m), 2.42-2.58 (2H, m), 2.75 (1H, quint, J = 7.6 Hz), 3.46 (1H, brs),
3.79 (1H, s), 4.09-4.27 (4H, m), 4.96 (2H, s), 5.27 (1H, s). 13 C NMR (CDCl 3
) (100 MHz): delta = 12.3, 13.97, 14.04, 24.4, 31.6, 36.1, 42.5, 45.6,
53.6, 55.5, 61.49, 61.53, 80.1, 120.7, 152.0, 167.7, 167.8. Ethyl [(RS,
5SR, 6SR) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL]
acetate 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.07
(3H, t, J = 7.6 Hz), 1.25 (3H, t, J = 7.6 Hz), 1.52 (1H, dd, J = 12.6,
7.2 Hz), 2.04 (1H, d, J = 16.4 Hz), 2.05-2.19 (2H, m), 2.23-2.35 (1H,
m), 2.50 (1H, dd, J = 15.8, 7.6 Hz), 2.62 (2H, s) , 2.86 (1H, quint, J =
7.6 Hz), 3.21 (1H, brs), 4.12 (4H, q, J = 7.6 Hz), 4.76 (2H, d, J =
11.6 Hz), 4.83 (2H, d, J = 11.6 Hz), 5.24 (1H, s).

(Example 16) [(RS, 5SR, 6RS)-3-ethyl-6- (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] propane diacid di -tert- butyl (racemic ) Di-tert-butyl [(RS, 5SR, 6RS) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] Propanedioate (Racemate)
[of 40] Di- tert-butyl [(1RS, 5SR) -3-ethylbicyclo [3.2.0]
hept-3-en-6-ylidene] propanedioate a (2.55 g) was dissolved in toluene
(26 mL), DBU (1.45 mL), nitromethane (1.05 mL) was added and stirred for
49 hours at room temperature. After quenching with 1 M HCl (50 mL), and
extracted three times with ethyl acetate (50 mL), and the resulting
ethyl acetate solution was washed with saturated brine (50 mL). The
solvent was distilled off under reduced pressure to give the title
compound as a pale yellow oil (2.36 g, 78% yield). 1 H NMR (CDCl 3
) (500 MHz): delta = 1.09 (t, 3H, J = 7.4 Hz), 1,45 (s, 9H), 1.49 (s,
9H), 1.71 (dd, 1H, J = 12.9, 7.4 Hz), 2.03 (d, 1H, 16.7 Hz), 2.09-2.19
(m, 2H), 2.47 (dd, 2H, J = 16.7, 7.9 Hz), 2.59 (ddd, 1H, J = 11.7, 8.9 ,
2.7 Hz), 2.67 (quint, 1H, J = 7.4 Hz), 3.52 (brs, 1H), 3.64 (s, 1H),
4.88 (d, 1H, J = 10.9 Hz), 4.95 (d, 1H, J = 10.9 Hz), 5.28 (m, 1H).

(Example 17) [(RS, 5SR, 6SR)-3-ethyl-6- (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] optical resolution of acetic acid [(1RS, 5SR, 6SR ) -3-Ethyl-6- (nitromethyl) bicyclo [3.2.0] optical resolution of hept-3-en-6-YL] acetic acid [of 41] [(RS, 5SR, 6SR) -3-Ethyl-6 - (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] acetic acid (0.2 g, 0.84 mmol) and CH 3
CN (3.0 mL) to dissolve the table of the optically active organic amine
of the following (0.42 mmol) was at room temperature stirred with,
precipitated filtered crystals selectivity and dried to determine yield.
The results I shown in the table below.[Table 1] * (1S, 5R, 6R) - the
body is the main product ** (1R, 5S, 6S) - the body is the main
product

(HPLC optical analysis condition)
Column: CHIRALPAK AD-RH 4.6 × 250 mm
mobile phase: 10 mM pH 2.0 phosphate buffer / MeCN = 25/75 (isocratic)
flow rate: 1.0 mL / min
Column temperature: 40 ° C
Detection wavelength: UV 210 nm
analysis time: 80 minutes
retention time: (1S, 5R, 6R) - Body: 35.2 min, (1R, 5S, 6S) - Body: 42.1 min
Column: CHIRALPAK AD-RH 4.6 × 250 mm
mobile phase: 10 mM pH 2.0 phosphate buffer / MeCN = 25/75 (isocratic)
flow rate: 1.0 mL / min
Column temperature: 40 ° C
Detection wavelength: UV 210 nm
analysis time: 80 minutes
retention time: (1S, 5R, 6R) - Body: 35.2 min, (1R, 5S, 6S) - Body: 42.1 min
(Example 18) [(1R, 5S, 6S)-3-ethyl-6- (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] acetic acid [(1R, 5S, 6S) -3 -Ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] acetic acid
[of 42] quinine (5.97 g, 18.4 mmol) was dissolved in acetone (300 mL),
[( RS, 5SR, 6SR) -3-Ethyl-6 (Nitromethyl) -bicyclo [3.2.0]
hept-3-en-6-YL] acetic acid (10.0 g, I was added 33.4 mmol). After
stirring 20 hours at room temperature, it was carried out 5 hours of
stirring it was cooled to 0 ° C. After filtering off the solid, washed
with cold acetone, the combined filtrate and washing was concentrated
under reduced pressure, further CH 3 CN were added and again
concentrated to the concentration residue (6.4 g, ee 65.2%) was
obtained. The resulting residue (6.4 g, ee 65.2%) and CH 3
was dissolved in CN (43 mL), (S) - it was added phenylglycinol (1.37 g, 1
eq minute) - (+). After stirring for 20 hours at room temperature and
stirred for 5 hours and cooled to 0 ° C. The precipitated crystals were
collected by filtration, and added to dilute hydrochloric acid and ethyl
acetate was dissolved by liquid separation, and dried under reduced
pressure after the organic layer was concentrated to give the title
compound (1.39 g, 14%, ee 92.0%). 1 H NMR (400 MHz, CDCl 3
): delta = 1.09 (t, 3H, J = 7.6 Hz), 1.47-1.57 (M 2H), 2.06-2.17 (M,
3H), 2.27-2.33 (M, 1H) , 2.49-2.55 (m, 1H), 2.66 (s, 2H),, 2.88 (quint,
1H, J = 7.6 Hz), 3.17 (bs, 1H), 4.78 (d, 1H, J = 11.5 Hz), 4.86 (d, 1H, J
= 11.5Hz), 5.27-5.28 (m, 1H)

(Example 28) [(1R, 5S, 6S)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid benzyl amine salt Benzylammonium [(1R, 5S, 6S) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetate
[of 52] Diethyl obtained by the method of Example 12 [(1RS, 5SR, 6RS)
-6-cyano -3-Ethylbicyclo [3.2.0] Hept- 3-en-6-YL] After the addition of
EtOH (390 mL) to CPME solution of propanedioate, heating under reflux, 8
N aqueous solution of potassium hydroxide (6.9 mL, 55.07 mmol ) after
adding a total of 5 times every 1 hour, refluxed for 5 hours and
returned to room temperature. The addition of water (60 mL) and 8N
aqueous potassium hydroxide (24 mL) to the above EtOH solution, and
after stirring for 2 h at 26-27 ° C, under reduced pressure at an
external temperature of 40-45 ° C until 150 mL It was concentrated. To
remove the organic layer by water (180 mL) and toluene (90 mL) was added
for liquid separation. The resulting aqueous solution Toluene (150 mL)
added, cooled to, was added concentrated hydrochloric acid 42.5 mL at
2-9 ° C, the pH was adjusted to 1.4. By separation to remove the aqueous
layer was added toluene (300 mL) benzylamine (23.6 g, 220.28 mmol) and.
After stirring for 30 minutes at 44-46 ° C make the inoculation, and
concentrated under reduced pressure until 300 mL at 44-46 ° C. After
stirring overnight at 22-23 ° C, and crystals were filtered off. And
vacuum dried at 40 ° C, was obtained as a white crystalline title
compound 54.4 g (79.2% from (1R, 5S) -3-Ethylbicyclo [3.2.0]
hept-3-en-6-one) a.

(Example 33) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid [(1R, 5S, 6S) - 6 (aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid
[of 57] Benzylammonium [(1R, 5S, 6S) -6-cyano-3-Ethylbicyclo [3.2. 0]
hept-3-en-6-yl] acetate (40.0 g) in toluene (200 mL), was added 2 mol / L
hydrochloric acid (100 mL) at room temperature and dissolved. And
allowed to stand the solution to drain the aqueous layer to obtain an
organic layer. To the stirred addition of 10% aqueous sodium chloride
solution (about 100 mL), and the aqueous layer was removed after
standing. The solution of water (100 mL) was added to, was adjusted to
10.0 to pH added 8 mol / L aqueous potassium hydroxide solution (about
15.7 mL), the organic layer was removed to standing. The solution to
the sponge cobalt (10 g), 28% aqueous ammonia (13 mL), 2%
dimethylpolysiloxane / toluene solution (2 mL) was added and warmed to
40 ° C in a hydrogen gas pressure (0.45 MPa) It was stirred for 8
hours.After cooling to room temperature, filtering the reaction mixture
to remove the sponge cobalt. The sponge cobalt on the filter it was
washed with water (80 mL). The resulting solution was stirred for 0.5
hours added the activated carbon (4 g), to remove the charcoal by
filtration. The activated carbon on the filter it was washed with water
(60 mL). The solution I was adjusted to about pH 6.0 with concentrated
hydrochloric acid (about 32.7g) a. Then, after stirring for 0.5 hours
was added potassium chloride (55.0 g), and cooled to 0 ° C. The
resulting was filtered and crystals were washed with 20% brine cooled to
about 0 ° C (80 mL), and dried overnight in vacuum at 50 ° C to give
the title compound as white crystals (26.9 g, content 88.3 %, 88.7%
content in terms of yield).

(Example 34) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid [(1R, 5S, 6S) - 6 (aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid
[of 58] (R) -Phenylethanaminium [(1R, 5S, 6S) -6-cyano-3 ethylbicyclo
[3.2.0] hept-3-en-6-yl] acetate (35.9g, 99.2 mmol, 95.7% de, ee 99.2%)
in toluene (120 mL) and 1 mol / L hydrochloric acid (150 mL) was added ,
it was stirred. After removing the aqueous layer, the organic layer was
washed twice with water (120 mL), and concentrated. The obtained
residue in MTBE to (150 mL) and sponge nickel (10.1 g) was added, under
hydrogen pressure (approximately 4 atm) and stirred for 3 hours at room
temperature. The reaction of 2 mol / L aqueous potassium hydroxide
solution (72 mL) was added, After stirring for 30 minutes, a sponge
nickel was filtered off. It was washed with a filtration sponge nickel 2
mol / L potassium hydroxide solution (12 mL). After combining the
filtrate and washings, the organic layer was removed to obtain an
aqueous layer. The organic layer was re-extracted with 2M aqueous
potassium hydroxide solution. The matched aqueous layer was cooled,
after adjusting the pH adding concentrated hydrochloric acid (about 12
mL) to 7.5, and the mixture was stirred at 0 ° C for about 3 hours.
Filtered the precipitated crystals were washed with ice-cold water (24
mL), and dried under reduced pressure at 50 ° C, to give the title
compound (18.3g, 88%, 99.8% de) and.

(Example 35) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid one benzenesulfonate [(1R, 5S, 6S)-6-(aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid Monobenzenesulfonate
[of 59] MTBE (83 mL), acetone (4.0 mL), water ( with respect to a
mixture of 0.98 mL), at 0 ° C [(1R, 5S, 6S) -6- (Aminomethyl)
-3-ethylbicyclo [3.2.0] hept-3-en-6-yl] acetic acid ( 4.07 g, 19.5 mmol)
was added and stirred to form a slurry solution. This BsOH (3.08 g,
19.5 mmol) it was added acetone (10.1 mL) solution of. 0 ° After
stirring for 1 hour at C, and stirred for 2 hours and allowed to warm to
room temperature. Over 1 hour and gradually cooled to -10 ° C, and
stirred for 2.5 hours. The resulting was filtered crystals, after
washing with acetone and cooled to 0 ° C (12 mL), and by vacuum-dried at
40 ° C, as white crystals of the title compound was obtained (6.44 g,
90.1% ). Various spectrum data of the obtained title compound was almost
(extent the structure can be identified) coincides with (described in
Patent Documents 5 and 6) the known information. (Purity measurement
method -1) column: Cadenza CW-C18 (Imtakt, 3 μm, 4.6 mm × 150 mm), 40 ° C
detection wavelength: UV 205 nm mobile phase: MeCN: 5 mM ammonium
hydrogen carbonate aqueous solution = ten ninety -80: 20 (gradient)
(0-12 min: MeCN 10%, 12-27 min: MeCN 10 → 80%, 27-45 min: MeCN 80%,
45-50 min: MeCN 80 → 10%, 50- 60 min: MeCN 10%, 60 min: STOP)
measurement time: 60 min flow rate: 1.0 mL / min infusion sample
concentration: 5mg / mL sample injection volume: 2μL retention time:
the title compound (as free form): 12.5 min diastereoisomers Marr
(Compound X): 13.5 min double bond position isomer (compound XII or
XIII): 9.4 min, 9.6 min, 11.4 min

| Patent | Submitted | Granted |
|---|---|---|
| Bicyclic [gamma]-amino acid derivative [US7947738] | 2010-09-30 | 2011-05-24 |
| Optical Resolution Methods for Bicyclic Compounds Using Enzymes [US2015038738] | 2014-10-10 | 2015-02-05 |
| WO2015005298A1 * | Jul 8, 2014 | Jan 15, 2015 | Daiichi Sankyo Company,Limited | METHOD FOR PRODUCING OPTICALLY ACTIVE BICYCLIC γ-AMINO ACID DERIVATIVE |
CONSTRUCTION
References
- Vinik A, Rosenstock J, Sharma U, Feins K, Hsu C, Merante D, et al. Efficacy and safety of mirogabalin (DS-5565) for the treatment of diabetic peripheral neuropathic pain: a randomized, double-blind, placebo- and active comparator-controlled, adaptive proof-of-concept phase 2 study. Diabetes Care. 2014 Dec;37(12):3253-61. doi: 10.2337/dc14-1044. PMID 25231896
- Vinik A, Sharma U, Feins K, Hsu C, Merante D. DS-5565 for the Treatment Of Diabetic Peripheral Neuropathic Pain: Randomized, Double-Blind, Placebo- And Active Comparator-Controlled Phase II Study (S20.004) Neurology April 8, 2014; 82(10): Supplement S20.004
Tokyo, Japan – (February 4, 2015)
– Daiichi Sankyo Company, Limited (hereafter, Daiichi Sankyo) today
announced enrollment of the first patients in large-scale,
multi-national clinical programs evaluating the safety and efficacy of
investigational mirogabalin (DS-5565), the first preferentially
selective alpha-2 delta ligand. The phase 3 clinical program across Asia
includes the REDUCER (An Asian, phase 3, multicenter, RandomizEd,
Double-blind, placebo-controlled 14-week stUdy of DS-5565 in patients
with diabetiC pEripheral neuRopathic pain followed by a 52-week
open-label extension) study and the NEUCOURSE (An AsiaN, phasE 3,
mUltiCenter, randomized, dOUble-blind, placebo-contRolled 14-week study
of DS-5565 in patientS with postherpetic neuralgia followed by a 52-week
open-label Extension) study which will evaluate investigational
mirogabalin for the treatment of diabetic peripheral neuropathic pain
(DPNP) and postherpetic neuralgia (PHN), respectively. The phase 3
global ALDAY (A Randomized, Double-Blind, Placebo- and Active-Controlled
Study of DS-5565 in Patients with Pain Associated with Fibromyalgia)
clinical program is ongoing and will evaluate mirogabalin for the
treatment of pain associated with fibromyalgia in three identical
studies.
“Pain associated with the neurologic conditions of diabetic peripheral neuropathic pain, postherpetic neuralgia and fibromyalgia can be debilitating,” said Lesley Arnold, MD, Professor of Psychiatry and Behavioral Neuroscience and Director of the Women’s Health Research Program, University of Cincinnati and lead investigator of the ALDAY program. “New treatment options are needed to help people living with these neurologic conditions relieve and manage their chronic pain and hopefully, improve their function and quality of life.”
“We are pleased that our global clinical development program evaluating the efficacy and safety of mirogabalin continues to move forward and has progressed into phase 3,” said Mahmoud Ghazzi, MD, PhD, Executive Vice President and Global Head of Development for Daiichi Sankyo. “Daiichi Sankyo is committed to identifying and studying new medicines that could help improve the management of chronic pain for people with diabetic peripheral neuropathy, postherpetic neuralgia and pain associated with fibromyalgia.”
About the REDUCER and NEUCOURSE Phase 3 Clinical Studies
The REDUCER study will last 14 weeks and is being conducted at approximately 200 centers in Japan, Taiwan and Korea. The NEUCOURSE study will also last 14 weeks and is being conducted at approximately 200 centers in Japan, Taiwan, Korea, Singapore, Malaysia and Thailand. The studies will include about 750 patients each with either diabetic peripheral neuropathic pain or postherpetic neuralgia, respectively. The objectives of the double-blind studies are to evaluate safety and efficacy of mirogabalin by comparing change in the average daily pain score (ADPS) from baseline to Week 14 in patients receiving a total daily dose of either 15 mg, 20 mg or 30 mg of mirogabalin versus placebo. Both studies will be followed by one-year open-label extension studies to assess long-term safety and efficacy of mirogabalin. For more information on the REDUCER study in patients with diabetic peripheral neuropathic pain, please visit
https://www.clinicaltrials.gov/ct2/show/NCT02318706?term=Mirogabalin&rank=3.
For more information on the NEUCOURSE study in patients with postherpetic neuralgia, please visithttps://www.clinicaltrials.gov/ct2/show/NCT02318719?term=Mirogabalin&rank=1.
About the ALDAY Phase 3 Clinical Program
The ALDAY program is a large clinical phase 3 program evaluating mirogabalin for the treatment of pain associated with fibromyalgia, and includes three, randomized, double-blind, placebo- and active-controlled studies, and an open label safety study that will be carried out over the next three years. Approximately 4,000 patients with pain associated with fibromyalgia will be enrolled at approximately 800 clinical centers at more than 40 countries worldwide. The primary objective of the studies in the ALDAY program is to compare change in weekly ADPS from baseline to Week 13 in patients receiving a total daily dose of either 15 mg or 30 mg of mirogabalin versus placebo. Weekly ADPS is based on daily pain scores reported by the patient that best describes his or her worst pain over the previous 24 hours. The primary objective of the phase 3 open-label extension study is to assess the long-term safety of a total daily dose of mirogabalin 15 mg or mirogabalin 30 mg in patients with pain associated with fibromyalgia. For more information on the studies in the ALDAY program, please visit
https://clinicaltrials.gov/ct2/show/NCT02187471?term=DS5565&rank=1
https://clinicaltrials.gov/ct2/show/NCT02187471?term=ds-5565&rank=2
https://clinicaltrials.gov/ct2/show/NCT02146430?term=ds-5565&rank=3
For more information on the open-label extension study, please visithttps://clinicaltrials.gov/ct2/show/NCT02234583?term=ds-5565&rank=4
For patient recruitment or additional clinical study information, please visit http://www.aldaystudy.com/.
About Diabetic Peripheral Neuropathic Pain
Diabetic peripheral neuropathy is a disorder that causes nerve damage to the extremities and is one of the most common long-term complications of diabetes.1 Symptoms include sharp pains or increased sensitivity, numbness, loss of balance and coordination, tingling, burning, or prickling sensations, which typically worsen at night.1 Up to 50 percent of people with diabetes have peripheral neuropathy2 and it is estimated that between 11 and 26 percent of people with diabetes experience diabetic peripheral neuropathic pain (DPNP).3-6 However, DPNP is often undertreated and underreported.2
About Postherpetic Neuralgia
Postherpetic neuralgia is pain that occurs after recovering from shingles, an infection that is caused by the herpes zoster (chickenpox) virus. Pain from postherpetic neuralgia can range in severity, and is typically described as burning, sharp, or stabbing.7 Other symptoms include sensitivity to touch, itching, numbness, and in rare cases, muscle weakness or paralysis can occur.7 The risk of developing postherpetic neuralgia increases with age and it mainly affects people older than 60.7 Studies have shown that only half of all patients affected with the condition will be relieved from pain within a year.8 Most people will require more than one treatment to help ease the pain.7
About Fibromyalgia
Fibromyalgia is a chronic disorder that causes widespread muscle pain, generalized tender points and fatigue.9 Other common symptoms include sleep disturbances, morning stiffness, memory and thinking problems (sometimes called fibro fog), tingling in the hands and feet and headaches.9 Fibromyalgia is often misdiagnosed and suboptimally treated.10-17 The overall estimated prevalence of fibromyalgia is approximately two to three percent in the general population, with a higher prevalence in women.18-22 Pain that occurs with fibromyalgia has a substantial impact on the patient, and can be associated with societal and economic burdens.23-29
About Mirogabalin
Mirogabalin is an investigational drug that is currently being studied for the treatment of DPNP, PHN and pain associated with fibromyalgia. Mirogabalin is preferentially selective in regards to how it binds to α2δ-1 subunit, a protein that may help to regulate how the brain processes pain signals. It has a unique binding profile and long duration of action.30*,31
About Daiichi Sankyo
Daiichi Sankyo Group is dedicated to the creation and supply of innovative pharmaceutical products to address the diversified, unmet medical needs of patients in both mature and emerging markets. While maintaining its portfolio of marketed pharmaceuticals for hypertension, dyslipidemia and bacterial infections used by patients around the world, the Group has also launched treatments for thrombotic disorders and is building new product franchises. Furthermore, Daiichi Sankyo research and development is focused on bringing forth novel therapies in oncology and cardiovascular-metabolic diseases, including biologics. The Daiichi Sankyo Group has created a "Hybrid Business Model," to respond to market and customer diversity and optimize growth opportunities across the value chain. For more information, please visit: www.daiichisankyo.com.
“Pain associated with the neurologic conditions of diabetic peripheral neuropathic pain, postherpetic neuralgia and fibromyalgia can be debilitating,” said Lesley Arnold, MD, Professor of Psychiatry and Behavioral Neuroscience and Director of the Women’s Health Research Program, University of Cincinnati and lead investigator of the ALDAY program. “New treatment options are needed to help people living with these neurologic conditions relieve and manage their chronic pain and hopefully, improve their function and quality of life.”
“We are pleased that our global clinical development program evaluating the efficacy and safety of mirogabalin continues to move forward and has progressed into phase 3,” said Mahmoud Ghazzi, MD, PhD, Executive Vice President and Global Head of Development for Daiichi Sankyo. “Daiichi Sankyo is committed to identifying and studying new medicines that could help improve the management of chronic pain for people with diabetic peripheral neuropathy, postherpetic neuralgia and pain associated with fibromyalgia.”
About the REDUCER and NEUCOURSE Phase 3 Clinical Studies
The REDUCER study will last 14 weeks and is being conducted at approximately 200 centers in Japan, Taiwan and Korea. The NEUCOURSE study will also last 14 weeks and is being conducted at approximately 200 centers in Japan, Taiwan, Korea, Singapore, Malaysia and Thailand. The studies will include about 750 patients each with either diabetic peripheral neuropathic pain or postherpetic neuralgia, respectively. The objectives of the double-blind studies are to evaluate safety and efficacy of mirogabalin by comparing change in the average daily pain score (ADPS) from baseline to Week 14 in patients receiving a total daily dose of either 15 mg, 20 mg or 30 mg of mirogabalin versus placebo. Both studies will be followed by one-year open-label extension studies to assess long-term safety and efficacy of mirogabalin. For more information on the REDUCER study in patients with diabetic peripheral neuropathic pain, please visit
https://www.clinicaltrials.gov/ct2/show/NCT02318706?term=Mirogabalin&rank=3.
For more information on the NEUCOURSE study in patients with postherpetic neuralgia, please visithttps://www.clinicaltrials.gov/ct2/show/NCT02318719?term=Mirogabalin&rank=1.
About the ALDAY Phase 3 Clinical Program
The ALDAY program is a large clinical phase 3 program evaluating mirogabalin for the treatment of pain associated with fibromyalgia, and includes three, randomized, double-blind, placebo- and active-controlled studies, and an open label safety study that will be carried out over the next three years. Approximately 4,000 patients with pain associated with fibromyalgia will be enrolled at approximately 800 clinical centers at more than 40 countries worldwide. The primary objective of the studies in the ALDAY program is to compare change in weekly ADPS from baseline to Week 13 in patients receiving a total daily dose of either 15 mg or 30 mg of mirogabalin versus placebo. Weekly ADPS is based on daily pain scores reported by the patient that best describes his or her worst pain over the previous 24 hours. The primary objective of the phase 3 open-label extension study is to assess the long-term safety of a total daily dose of mirogabalin 15 mg or mirogabalin 30 mg in patients with pain associated with fibromyalgia. For more information on the studies in the ALDAY program, please visit
https://clinicaltrials.gov/ct2/show/NCT02187471?term=DS5565&rank=1
https://clinicaltrials.gov/ct2/show/NCT02187471?term=ds-5565&rank=2
https://clinicaltrials.gov/ct2/show/NCT02146430?term=ds-5565&rank=3
For more information on the open-label extension study, please visithttps://clinicaltrials.gov/ct2/show/NCT02234583?term=ds-5565&rank=4
For patient recruitment or additional clinical study information, please visit http://www.aldaystudy.com/.
About Diabetic Peripheral Neuropathic Pain
Diabetic peripheral neuropathy is a disorder that causes nerve damage to the extremities and is one of the most common long-term complications of diabetes.1 Symptoms include sharp pains or increased sensitivity, numbness, loss of balance and coordination, tingling, burning, or prickling sensations, which typically worsen at night.1 Up to 50 percent of people with diabetes have peripheral neuropathy2 and it is estimated that between 11 and 26 percent of people with diabetes experience diabetic peripheral neuropathic pain (DPNP).3-6 However, DPNP is often undertreated and underreported.2
About Postherpetic Neuralgia
Postherpetic neuralgia is pain that occurs after recovering from shingles, an infection that is caused by the herpes zoster (chickenpox) virus. Pain from postherpetic neuralgia can range in severity, and is typically described as burning, sharp, or stabbing.7 Other symptoms include sensitivity to touch, itching, numbness, and in rare cases, muscle weakness or paralysis can occur.7 The risk of developing postherpetic neuralgia increases with age and it mainly affects people older than 60.7 Studies have shown that only half of all patients affected with the condition will be relieved from pain within a year.8 Most people will require more than one treatment to help ease the pain.7
About Fibromyalgia
Fibromyalgia is a chronic disorder that causes widespread muscle pain, generalized tender points and fatigue.9 Other common symptoms include sleep disturbances, morning stiffness, memory and thinking problems (sometimes called fibro fog), tingling in the hands and feet and headaches.9 Fibromyalgia is often misdiagnosed and suboptimally treated.10-17 The overall estimated prevalence of fibromyalgia is approximately two to three percent in the general population, with a higher prevalence in women.18-22 Pain that occurs with fibromyalgia has a substantial impact on the patient, and can be associated with societal and economic burdens.23-29
About Mirogabalin
Mirogabalin is an investigational drug that is currently being studied for the treatment of DPNP, PHN and pain associated with fibromyalgia. Mirogabalin is preferentially selective in regards to how it binds to α2δ-1 subunit, a protein that may help to regulate how the brain processes pain signals. It has a unique binding profile and long duration of action.30*,31
About Daiichi Sankyo
Daiichi Sankyo Group is dedicated to the creation and supply of innovative pharmaceutical products to address the diversified, unmet medical needs of patients in both mature and emerging markets. While maintaining its portfolio of marketed pharmaceuticals for hypertension, dyslipidemia and bacterial infections used by patients around the world, the Group has also launched treatments for thrombotic disorders and is building new product franchises. Furthermore, Daiichi Sankyo research and development is focused on bringing forth novel therapies in oncology and cardiovascular-metabolic diseases, including biologics. The Daiichi Sankyo Group has created a "Hybrid Business Model," to respond to market and customer diversity and optimize growth opportunities across the value chain. For more information, please visit: www.daiichisankyo.com.
| trial(s) |
|
|---|
 | |
| Systematic (IUPAC) name | |
|---|---|
(1R,5S,6S)-6-(aminomethyl)-3-ethyl-bicyclo(3.2.0)hept-3-ene-6-acetic acid
| |
| Identifiers | |
| CAS Registry Number | 1138245-21-2 |
| PubChem | CID: 49802951 |
| ChemSpider | 32701007 |
| Chemical data | |
| Formula | C12H19NO2 |
| Molecular mass | 209.285 g/mol |
/////////
1138245-13-2, CCC1=C[C@@H]2[C@H](C1)C[C@@]2(CC(=O)O)CN
CCC1=CC2C(C1)CC2(CC(=O)O)CN
smiles besylate......CCC1=C[C@@H]2[C@H](C1)C[C@@]2(CC(=O)O)CN.c1ccc(cc1)S(=O)(=O)O
2 Pregabalin

PREGABALIN
LEARN MORE AT
http://orgspectroscopyint.blogspot.in/2013/11/pregabalin-spectral-data.html
Pregabalin (INN) /prɨˈɡæbəlɨn/ is an anticonvulsant drug used for neuropathic pain and as an adjunct therapy for partial seizures with or without secondary generalization in adults.[1] It has also been found effective for generalized anxiety disorder and is (as of 2007) approved for this use in the European Union and Russia. It was designed as a more potent successor to gabapentin. Pregabalin is marketed by Pfizer under the trade nameLyrica. Pfizer described in an SEC filing that the drug could be used to treat epilepsy, post-herpetic neuralgia, diabetic peripheral neuropathy and fibromyalgia. Sales reached a record $3.063 billion in 2010. In Bangladesh Pregabalin is sold under the brand of Nervalin by Beximco Pharma It is effective at treating some causes of chronic pain such as fibromyalgia but not others. It is considered to have a low potential for abuse, and a limited dependence liability if misused, but is classified as a Schedule V drug in the U.S.
LEARN MORE AT
http://orgspectroscopyint.blogspot.in/2013/11/pregabalin-spectral-data.html
Pregabalin (INN) /prɨˈɡæbəlɨn/ is an anticonvulsant drug used for neuropathic pain and as an adjunct therapy for partial seizures with or without secondary generalization in adults.[1] It has also been found effective for generalized anxiety disorder and is (as of 2007) approved for this use in the European Union and Russia. It was designed as a more potent successor to gabapentin. Pregabalin is marketed by Pfizer under the trade nameLyrica. Pfizer described in an SEC filing that the drug could be used to treat epilepsy, post-herpetic neuralgia, diabetic peripheral neuropathy and fibromyalgia. Sales reached a record $3.063 billion in 2010. In Bangladesh Pregabalin is sold under the brand of Nervalin by Beximco Pharma It is effective at treating some causes of chronic pain such as fibromyalgia but not others. It is considered to have a low potential for abuse, and a limited dependence liability if misused, but is classified as a Schedule V drug in the U.S.
Lyrica
is one of four drugs which a subsidiary of Pfizer in 2009 pleaded
guilty to misbranding "with the intent to defraud or mislead". Pfizer
agreed to pay $2.3 billion (£1.4 billion) in settlement, and entered a
corporate integrity agreement. Pfizer illegally promoted the drugs and
caused false claims to be submitted to government healthcare programs
for uses that were not approved by the U.S. Food and Drug Administration (FDA
In the United States, the Food and Drug Administration (FDA) has approved pregabalin for adjunctive therapy for adults with partial onset seizures, management of postherpetic neuralgia andneuropathic pain associated with spinal cord injury and diabetic peripheral neuropathy, and the treatment of fibromyalgia. Pregabalin has also been approved in the European Union and Russia(but not in US) for treatment of generalized anxiety disorder.


READ AT ...............http://www.rsc.org/chemistryworld/News/2008/July/09070801.asp

- (S)-Pregabalin, (S)-(+)-3-(aminomethyl)-5-methylhexanoic acid, a compound having the chemical structure,
is also known as γ-amino butyric acid or (S)-3-isobutyl GABA. (S)-Pregabalin, marketed under the name LYRICA®, has been found to activate GAD (L-glutamic acid decarboxylase). (S)-Pregabalin has a dose dependent protective effect on-seizure, and is a CNS-active compound. (S)-Pregabalin is useful in anticonvulsant therapy, due to its activation of GAD, promoting the production of GABA, one of the brain's major inhibitory neurotransmitters, which is released at 30 percent of the brains synapses. (S)-Pregabalin has analgesic, anticonvulsant, and anxiolytic activity. - [0003]Several processes for the synthesis of (S)-Pregabalin are known. For example, see DRUGS OF THE FUTURE, 24 (8), 862-870 (1999). One such process is illustrated in scheme 1.
- [0004]In Scheme 1, 3-isobutyl glutaric acid, compound 2, is converted into the corresponding anhydride, compound 3, by treatment with refluxing acetic anhydride. The reaction of the anhydride with NH4OH produces the glutaric acid mono-amide, compound 4, which is resolved with (R)-1-phenylethylamine, yielding the (R)-phenylethylamine salt of (R)-3-(carbamoylmethyl)-5-methylhexanoic acid, compound 5. Combining the salt with an acid liberates the R enantiomer, compound 6. Finally, a Hoffmann degradation with Br2/NaOH provides (S)-Pregabalin. A disadvantage of this method is that it requires separating the two enantiomers, thereby resulting in the loss of half the product, such that the process cost is high.
- [0005]Several stereoselective processes for the synthesis of (S)-Pregabalin have been disclosed. For example, U.S. Patent No. 5,599,973 discloses the preparation of (S)-Pregabalin using stoichiometric (+)-4-methyl-5-phenyl-2-oxazolidinone as a chiral auxiliary that may be recycled. In general, however, that route is of limited use for scale-up, principally due to the low temperature required for the reactions, the use of pyrophoric reagent, such as, butyl lithium, to side reactions, and due to a low overall yield.
- [0006]Another process is disclosed inU.S. Patent Application Publication No. 2003/0212290 , which discloses asymmetric hydrogenation of a cyano-substituted olefin, compound 7, to produce a cyano precursor of (S)-3-(aminomethyl)-5-methyl hexanoic acid, compound 8, as seen in scheme 2.
- [0007]Subsequent reduction of the nitrile in compound 8 by catalytic hydrogenation produces (S)-Pregabalin. The cyano hexenoate starting material, compound 7, is prepared from 2-methyl propanal and acrylonitrile (Yamamoto et al, Bull. Chem. Soc. Jap., 58, 3397 (1985)). However, the disclosed method requires carbon monoxide under high pressure, raising serious problems in adapting this scheme for production scale processes.
- [0008]A process published by G.M. Sammis, et al., J. Am. Chem. Soc., 125(15), 4442-43 (2003), takes advantage of the asymmetric catalysis of cyanide conjugate addition reactions. The method discloses the application of aluminum salen catalysts to the conjugate addition of hydrogen cyanide to α,β-unsaturated imides as shown in scheme 3. Reportedly, TMSCN is a useful source of cyanide that can be used in the place of HCN. Although the reaction is highly selective, this process is not practicable for large scale production due to the use of highly poisonous reagents. Moreover, the last reductive step requires high pressure hydrogen, which only adds to the difficulties required for adapting this scheme for a production scale process.
- [0009]In 1989, Silverman reported a convenient synthesis of 3-alkyl-4-amino acids compounds in SYNTHESIS, Vol. 12, 953-944 (1989). Using 2-alkenoic esters as a substrate, a series of GABA analogs were produced by Michael addition of nitromethane to α,β-unsaturated compounds, followed by hydrogenation at atmospheric pressure of the nitro compound to amine moiety as depicted in scheme 4.
- [0010]Further resolution of compound 14 may be employed to resolve Pregabalin. This, of course, results in the loss of 50 percent of the product, a serious disadvantage. However, the disclosed methodology reveals that the nitro compound can serve as an intermediate for the synthesis of 3-alkyl-4-amino acids.
- [0011]Recent studies have indicated that cinchona alkaloids are broadly effective in chiral organic chemistry. A range of nitroalkenes were reportedly treated with dimethyl or diethyl malonate in THF in the presence of cinchona alkaloids to provide high enantiomeric selectivity of compound 15,
and its analogues. For example, see H. Li, et al., J. Am. Chem. Soc, 126(32), 9906-07 (2004). These catalysts are easily accessible from either quinine or quinidine, and are reportedly highly efficient for a synthetically C-C bond forming asymmetric conjugate addition as shown in scheme 5.
- [0012]R3 represents several alkyl and aryl groups. The scope of the reaction has been extended to other nitroolefins and applied to prepare ABT-546 employing bis(oxazoline)Mg(OTf)2. See, for example, D.M. Barnes, et al., J. Am. Chem. Soc., 124(44), 13097-13105 (2002).
- [0013]Other groups have investigated a new class of bifunctional catalysts bearing a thiourea moiety and an amino group on a chiral scaffold. SeeT. Okino, et al., J. Am. Chem. Soc., 127(1), 119-125 (2005). On the basis of a catalytic Michael addition to the nitroolefin with enantiomeric selectivity, they were able to prepare a series of analogues of compound 15.
- [0014]Thus, there is a need in the art for new processes for the preparation of (S)-Pregabalin that does not suffer from the disadvantages mentioned above. Chemical Abstracts, database accession no. 2005:236589 refers to a process for the synthesis of pregabalin using methyl cyanoacetate, by condensation, addition, cyclization, aminolysis, Hoffmann rearrangement and resolution with (S)-mandelic acid.
Karenewsky, D. S., et al., J. Org. Chem., 1991, 56, 3744-3747, discloses reaction of a glutaric acid anhydride with (S)-1-phenyethylamine to prepare the corresponding amide, which is subsequently used to prepare β-ketophosphonate derivatives.
Verma, R., et al., J. Chem. Soc. Perkin Trans. I, 1999, 257-264, discloses desymmetrization of prochiral anhydrides with Evans' oxazolidinones to prepare homochiral glutaric and adipic acid derivatives.
Shintani, R. et al., Angew. Chem. Int. Ed. 2002, 41 (6), 1057-1059, discloses the desymmetrization of various glutaric acid anhydrides using Grignard reagents in the presence of (-)-sparteine
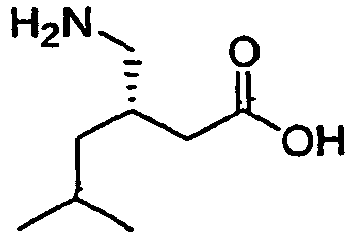








Pregabalin
(Lyrica®) is a lipophilic GABA analogue that is prescribed for the
treatment of epilepsy. This short, small-scale synthesis of pregabalin
features a highly enantioselective asymmetric conjugate addition of the
alkenyl trifluoroborate B to the α,β-unsaturated lactam A catalyzed by a rhodium complex incorporating the chiral bicyclo[3.3.0]octa-2,5-diene ligand L.
A
further 17 examples of this new variant of the Hayashi–Miyaura
asymmetric conjugate addition reaction are reported using six
α,β-unsaturated carbonyl substrates and ten alkenyl trifluoroborates.
The asymmetric conjugate addition was also applied to the synthesis of
the potent neuroexcitatory agent α-kainic acid (seven steps, 40% overall
yield).



fruits from Rosaceae family (Genus: Prunus). The collected fruits are peach (Prunus persica), Himalayan wild cherry (Prunus avium), Red Indian plum (Prunusdomestica), Himalayan plum (Prunus americana), apricot (Prunus armeniaca) and shakarpara (white apricot, a hybrid cultivar of normal apricot found in Nepal and India). All the six newly found HNLs are R-selective, i.e., they yield R-mandelonitrile from benzaldehyde bySi-facial attack of the cyanide anion. The enantioselectivity obtained for the formation of mandelonitriles by all the six HNLs are in the range of 60-93%. The best results are obtained with PavHNL and ParsHNL (both provide 93% ee), where as PpHNL is the least enantioselective (provides 60% ee for R-mandelonitrile formation). The main object of the project proposal will be the development of efficient biocatalytic route for various value added products such as Pregablin, Baclofen and Pril drugs

http://www.google.com/patents/EP2053040A1

http://www.google.com/patents/EP2170813A2

http://www.google.com/patents/EP2170813A2
fruits from Rosaceae family (Genus: Prunus). The collected fruits are peach (Prunus persica), Himalayan wild cherry (Prunus avium), Red Indian plum (Prunusdomestica), Himalayan plum (Prunus americana), apricot (Prunus armeniaca) and shakarpara (white apricot, a hybrid cultivar of normal apricot found in Nepal and India). All the six newly found HNLs are R-selective, i.e., they yield R-mandelonitrile from benzaldehyde bySi-facial attack of the cyanide anion. The enantioselectivity obtained for the formation of mandelonitriles by all the six HNLs are in the range of 60-93%. The best results are obtained with PavHNL and ParsHNL (both provide 93% ee), where as PpHNL is the least enantioselective (provides 60% ee for R-mandelonitrile formation). The main object of the project proposal will be the development of efficient biocatalytic route for various value added products such as Pregablin, Baclofen and Pril drugs
http://www.google.com/patents/EP2053040A1
http://www.google.com/patents/EP2170813A2
http://www.google.com/patents/EP2170813A2
 LEARN MORE AT
LEARN MORE AThttp://orgspectroscopyint.blogspot.in/2013/11/pregabalin-spectral-data.html
3 ATAGABALIN

Atagabalin
Trans-dimethyl gababutin; UNII-JT7957Q2FB; 223445-75-8;2-[(3S,4S)-1-(aminomethyl)-3,4-dimethylcyclopentyl]acetic acid
DNC014878
AN-5147
PD-0200390
D09581
2-[(3S,4S)-1-(aminomethyl)-3,4-dimethyl-cyclopentyl]acetic acid
3,4-trans-2-(1-(aminomethyl)-3,4-dimethylcyclopentyl)acetic acid
Cyclopentaneaceticacid, 1-(aminomethyl)-3,4-dimethyl-, (3S,4S)-
Pfizer Inc. INNOVATOR
Atagabalin (PD-0200,390) is a drug developed by Pfizer and related to gabapentin, which similarly binds to the α2δ calcium channels (1 and 2).[1] It was under development as a treatment for insomnia,[2][3][4] but was discontinued following unsatisfactory trial results.
Gabapentin (Neurontin®) (1) was
launched as an add-on therapy for epilepsy in 1994. Utility against
neuropathic pain and anxiety have been reported preclinically and
efficacy against neuropathic pain has been demonstrated clinically in
humans. Pregabalin (Lyrica®) (2),
has superior potency and pharmacokinetics to gabapentin and has been
approved for the management of neuropathic pain associated with diabetic
peripheral neuropathy, post-herpetic neuralgia, adjunctive treatment of
partial seizures, and fibromyalgia in the US.


SYNTHESIS

PATENT
WO 1999021824http://www.google.co.in/patents/WO1999021824A1?cl=en
synthesis of 3-oxo-2,8-diazaspiro[4,5]decane-
8-carboxylic acid tert-butyl ester (P. W. Smith et al., J. Med. Chem., 1995;38:3772). The compounds may also be synthesized by the methods outlined by G. Satzinger et al., (Ger Offen 2,460,891; US 4,024,175, and Ger Offen 2,611,690; US 4,152,326) (General Schemes 3 and 4). The compounds may also be synthesized by the route outlined by G. Griffiths et al., Helv. Chim. Ada, 1991 ;74:309 (General Scheme 5). General Scheme 1

General Scheme 2


General Scheme 3

General Scheme 4

General Scheme 5


EXAMPLE 1
NaH (60% dispersion in oil, 737 mg, 18.42 mmol) was suspended in dry tetrahydrofuran (50 mL) and cooled to 0°C. Triethylphosphonoacetate (3.83 mL, 19.30 mmol) was added and the mixture stirred at 0°C for 15 minutes. The ketone (1) (1.965 g, 17.54 mmol) in THF (10 mL) was then added and the mixture allowed to warm to room temperature. After 2 hours, the mixture was partitioned between diethyl ether (200 mL) and water (150 mL). The organic phase was separated, washed with brine, dried (MgSO4) and the solvent removed in vacuo.
The residue was purified by flash chromatography (silica, ethyl acetate:heptane 1 :9) to give 3.01 g (94%) of (2) as a colorless oil.
*H NMR 400 MHz (CDCI3): δ 1.01 (3H, d, J = 6 Hz), 1.03 (3H, d, J = 6 Hz), 1.26
(3H, t, J = 7 Hz), 1.49 (2H, m), 2.07 (1H, m), 2.24 (1H, m), 2.61 (1H, m), 4.13 (2H, q, J = 7 Hz), 5.72 (1H, s).
MS (CI+) m/e: 183 ([MH+], 18%).
Synthesis of (trans)-(3,4-Dimethyl-l-nitromethyl-cyclopentyl)-acetic acid ethyl ester (3)
The unsaturated ester (2) (2.95 g, 16.2 mmol) was dissolved in tetrahydrofuran (10 mL) and stirred at 70°C with nitromethane (1.9 mL, 35.2 mmol) and tetrabutylammonium fluoride (1.0 M in tetrahydrofuran, 22 mL, 22.0 mmol). After 6 hours, the mixture was cooled to room temperature, diluted with ethyl acetate (50 mL), and washed with 2N HCl (30 mL) followed by brine (50 mL). The organic phase was collected, dried (MgSO4) and the solvent removed in vacuo. The residue was purified by flash chromatography (silica, ethyl acetate :heptane 1 :9) to give 1.152 g (29%) of a clear oil. !H NMR 400 MHz (CDCI3): δ 0.98 (6H, d, J = 6 Hz), 1.10-1.39 (5H, m), 1.47
(2H, m), 1.87 (1H, m), 2.03 (1H, m), 2.57 (2H, ABq, J = 16, 38 Hz), 4.14 (2H, q, J = 7 Hz), 4.61 (2H, ABq, J = 12, 60 Hz).
MS (ES+) m/e: 244 ([MH+], 8%).
IR (film) v ein-1 : 1186, 1376, 1549, 1732, 2956. Synthesis of (±)-(trans)-7,8-Dimethyl-spiro[4.4]nonan-2-one (4)
The nitroester (3) (1.14 g, 4.7 mmol) was dissolved in methanol (50 mL) and shaken over Raney nickel catalyst under an atmosphere of hydrogen (40 psi) at 30°C. After 5 hours, the catalyst was removed by filtration through celite. The solvent was removed in vacuo to give 746 mg (95%) of a pale yellow oil which solidified on standing.
! H NMR 400 MHz (CDC13): δ 0.98 (6H, d, J = 6 Hz), 1.32 (2H, m), 1.46 (2H, m), 1.97 (2H, m), 2.27 (2H, ABq, J = 16, 27 Hz), 3.23 (2H, s), 5.62 (1H, br s). MS (ES+) m/e: 168 ([MH+], 100%). IR Cfilπ v cm-1 : 1451, 1681, 1715, 2948, 3196.
Synthesis of (±)-(trans)-(l-Aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid hydrochloride (5)
The lactam (4) (734 mg, 4.40 mmol) was heated to reflux in a mixture of 1 ,4-dioxan (5 mL) and 6N HCl (15 mL). After 4 hours, the mixture was cooled to room temperature, diluted with water (20 mL), and washed with dichloromethane
(3 x 30 mL). The aqueous phase was collected and the solvent removed in vacuo. The residue was triturated with ethyl acetate to give 675 mg (69%) of a white solid after collection and drying.
ΪH NMR 400 MHz (d6-DMSO): δ 0.91 (6H, d, J = 6 Hz), 1.18 (2H, m), 1.42 (2H, m), 1.72 (1H, m), 1.87 (1H, m), 2.42 (2H, ABq, J = 16, 24Hz), 2.90 (2H, ABq,
J = 12, 34 Hz), 8.00 (3H, br s), 12.34 (1H, br s).
MS (ES+) m/e: 186 ([MH-HC1J+, 100%).
PATENT
WO 2002000209PATENT
http://www.google.co.in/patents/WO1999021824A1?cl=enPATENT
WO 2007010387http://www.google.com/patents/WO2007010387A2?cl=en


PAPER
Synthesis and in vivo evaluation of 3,4-disubstituted gababutinsBioorganic&Medicinal Chemistry Letters (2010), 20, (1), 248-251.
The synthesis of 3,4-trans-dimethyl cyclopentanone (14), is detailed in Scheme 1.
-
Scheme 1.
Reagents and conditions: (i) (−)-menthol, pyridine, CH2Cl2; (ii) butadiene, TiCl4, toluene, −10 °C (100% yield, 65% de) or butadiene, Et2AlCl, toluene, −60 °C (64% yield, 95% de); (iii) LiAlH4, THF; recrystallisation from acetone; (iv) pyridine, MsCl, 0 °C, 18h (82%); (v) LiAlH4, diethyl ether, 40 °C, 2h (98%); (vi) KMnO4, nBu4NBr, H2O–CH2Cl2, rt, 18h; then SO2, 0 °C (82%); (vii) methanol, cH2SO4, rt, 18h (90%) (viii) KOtBu, THF, 75 °C, 3h (100%); (ix) DMSO, H2O, 140 °C, 4 h (86%).
-
Scheme 3.
Reagents and conditions: (i) triethylphosphonoacetate, NaH, THF, 0 °C to rt (95%); (ii) MeNO2, TBAF, THF, reflux (65%); (iii) H2, Ni, MeOH; (iv) 6 N HCl, 1,4-dioxane, reflux (69% from nitroester).


References
1 Blakemore
DC, Bryans JS, Carnell P, Carr CL, Chessum NE, Field MJ, Kinsella N,
Osborne SA, Warren AN, Williams SC (January 2010). “Synthesis and in
vivo evaluation of bicyclic gababutins”. Bioorganic & Medicinal Chemistry Letters 20 (2): 461–4. doi:10.1016/j.bmcl.2009.11.118. PMID 20005103.
- 2 Corrigan B, Feltner DE, Ouellet D, Werth JL, Moton AE, Gibson G (August 2009). “Effect of renal impairment on the pharmacokinetics of PD 0200390, a novel ligand for the voltage-gated calcium channel alpha-2-delta subunit”. British Journal of Clinical Pharmacology 68 (2): 174–80. doi:10.1111/j.1365-2125.2009.03444.x. PMC 2767279. PMID 19694735.
- 3 Quintero JE, Pomerleau F, Huettl P, Johnson KW, Offord J, Gerhardt GA (May 2011). “Methodology for rapid measures of glutamate release in rat brain slices using ceramic-based microelectrode arrays: Basic characterization and drug pharmacology”. Brain Research 1401: 1–9. doi:10.1016/j.brainres.2011.05.025. PMID 21664606.
- 4 Kjellsson MC, Ouellet D, Corrigan B, Karlsson MO (June 2011). “Modeling Sleep Data for a New Drug in Development using Markov Mixed-Effects Models”. Pharmaceutical Research 28 (10): 2610–27. doi:10.1007/s11095-011-0490-x. PMID 21681607.
| Patent | Submitted | Granted |
|---|---|---|
| Pyrazolo[4,3-d]pyrimidines as Phosphodiesterase Inhibitors [US7572799] | 2005-11-03 | 2009-08-11 |
| Substituted morpholine compounds for the treatment of central nervous system disorders [US7659394] | 2005-11-03 | 2010-02-09 |
| Therapeutic pyrazolo[3,4-B]pyridines and indazoles [US7423054] | 2006-06-01 | 2008-09-09 |
| Amide derivatives as ion-channel ligands and pharmaceutical compositions and methods of using the same [US7312233] | 2006-09-14 | 2007-12-25 |
| Compounds useful in therapy [US7482375] | 2006-10-26 | 2009-01-27 |
| Therapeutic pyrazolo[3,4-b]pyridines and indazoles [US7485636] | 2006-09-28 | 2009-02-03 |
| Substituted N-sulfonylaminophenylethyl-2-phenoxyacetamide compounds as VR1 receptor antagonists [US7566739] | 2006-09-14 | 2009-07-28 |
| Amide derivatives as ion-channel ligands and pharmaceutical compositions and methods of using the same [US7576099] | 2006-08-31 | 2009-08-18 |
| Substituted sulfonylaminoarylmethyl cyclopropanecarboxamide as VR1 receptor antagonists [US7622589] | 2006-09-21 | 2009-11-24 |
| Alpha 2 Delta Ligands for Fibromyalgia and Other Disorders [US2009203782] | 2009-08-13 |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
[(3S,4S)-1-(aminomethyl)-3,4-dimethylcyclopentyl]acetic acid
|
|
| Identifiers | |
| CAS Registry Number | 223445-75-8 |
| ATC code | None |
| PubChem | CID: 9794485 |
| ChemSpider | 7970252 |
| UNII | JT7957Q2FB |
| ChEMBL | CHEMBL593430 |
| Chemical data | |
| Formula | C10H19NO2 |
| Molecular mass | 185.263 g/mol |
4 COMING................
Very Informative content on CaryophylleneThank you for the article!
ReplyDelete