| Molecular Formula: | C31H35F7N4O2 |
|---|---|
| Molecular Weight: | 628.624022 g/mol |
CAS 579475-18-6
Orvepitant (GW823296)
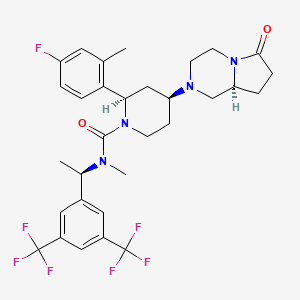
(2R,4S)-4-[(8aS)-6-oxo-1,3,4,7,8,8a-hexahydropyrrolo[1,2-a]pyrazin-2-yl]-N-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-2-(4-fluoro-2-methylphenyl)-N-methylpiperidine-1-carboxamide

MALEATE
CAS [579475-24-4] MALEATE
MF C31H35F7N4O2.C4H4O4
MW 744.70
MW 744.70
- Phase IICough; Pruritus
- DiscontinuedAnxiety disorders; Major depressive disorder; Post-traumatic stress disorders
Most Recent Events
- 19 Dec 2015NeRRe Therapeutics terminates a phase II trial in Pruritus in Italy and the United Kingdom (EudraCT2013-002763-25)
- 16 Dec 2013No development reported - Phase-II for Post-traumatic stress disorder in USA (PO)
- 16 Dec 2013No development reported - Phase-II for Major depressive disorder in Canada (PO)
| Latest Stage of Development | Phase II |
| Standard Indication | Itch |
| Indication Details | Treat intense pruritus (itch) associated with epidermal growth factor receptor inhibitor (EGFRi) anticancer therapies |
Start of Phase II study of neurokinin-1 receptor antagonist orvepitant for intense pruritus induced by epidermal growth factor receptor inhibitors
First Clinical Trial for NeRRe Therapeutics
Stevenage, UK, 23 January 2014.NeRRe Therapeutics Ltd, which is focused on the development of neurokinin (NK) receptor antagonists for a range of indications, is pleased to announce the start of a Phase II study of the novel NK-1 receptor antagonist orvepitant. The proof-of-concept study, results of which are expected in 2015, is investigating orvepitant’s effectiveness as a treatment for the intense pruritus (itch) associated with epidermal growth factor receptor inhibitor (EGFRi) anticancer therapies. The itch intensity experienced by patients can be so severe that their EGFRi dose must be reduced or the treatment withdrawn; also pruritus along with rash has a significant effect on quality of life1.
The RELIEVE-1 trial is a randomised, double-blind, placebo-controlled study to evaluate the safety, tolerability and efficacy of two daily dose levels of oral orvepitant on EGFRi-induced intense pruritus in oncology subjects. Its primary endpoint is the difference between orvepitant and placebo in reducing the intensity of pruritus over 4 weeks, as measured on a subject-recorded numerical rating scale. RELIEVE-1 is being undertaken in 15 clinical sites in Italy, with Dr Bruno Vincenzi from Università Campus Bio-Medico di Roma as lead investigator. Dr Vincenzi and his colleagues at the centre have pioneered the use NK-1 antagonists as anti-pruritics in this setting2. Chemistry, manufacturing and control support for RELIEVE-1 is being provided by Aptuit (Verona) Srl, with clinical operations assistance from the CRO Cromsource.
Dermatologic adverse events such as pruritus are a common feature of targeted anti-cancer therapies, with incidence of this symptom induced by EGFRia drugs in clinical trials ranging from 14.6% to 54.9% depending on the specific agent3. Open-label studies in patients suffering from refractory chronic pruritus have indicated that NK-1 receptor antagonism can provide rapid and highly effective relief as well as significantly improving quality of life.2,4,5,6
Dr Mike Trower, Co-founder & Chief Operating Officer of NeRRe Therapeutics said:
‘We are very pleased to announce the start of RELIEVE-1, NeRRe’s first clinical trial, in this important area of unmet medical need. There is a strong rationale and a growing body of clinical evidence supporting the potential of orvepitant as an anti-pruritic for this devastating symptom commonly associated with EGFRis. Given its known effects on mood and sleep, orvepitant may also provide additional benefits for patient well-being.’
Dr Emiliangelo Ratti, NeRRe Therapeutics Co-founder added:
‘The intense pruritus induced by EGFRis can lead to significant suffering and poor quality of life, and we believe that a treatment for this troubling side effect would be welcomed by cancer patients and supportive care doctors alike. A successful study of orvepitant in this indication would provide further evidence of the broad therapeutic potential of the NK-1 receptor antagonist mechanism which NeRRe is exploiting in its pipeline.’
–ENDS–
a This includes monoclonal antibodies that target the extracellular domain of EFGR, small molecule tyrosine kinase (TK) inhibitors, and small molecule dual TK inhibitors.About NeRRe Therapeutics
NeRRe Therapeutics was formed in December 2012 and is focussed on the development of a portfolio of NK receptor antagonists acquired from GlaxoSmithKline (GSK), which have therapeutic potential in a broad range of indications. NeRRe Therapeutics was co-founded by Drs Emiliangelo Ratti and Mike Trower, both of whom are both former senior leaders of neurosciences drug discovery at GSK with intimate knowledge of the transferred assets and the neurokinin receptor system field. In 2012 NeRRe Therapeutics raised £11.5 million ($18.4 million) in Series A financing from two leading European financial institutions, Novo A/S (www.novo.dk/ventures) and Advent Life Sciences (www.adventventures.com), who are represented by Dr Martin Edwards (Chairman) and Dr Kaasim Mahmood respectively on the company’s Board.
NeRRe (www.nerretherapeutics.com) is based at the state-of-the-art Stevenage Bioscience Catalyst (www.stevenagecatalyst.com), the UK’s first open innovation bioscience campus.
About Orvepitant
Orvepitant is a ‘novel generation’ brain penetrant, selective and potent, small molecule NK-1 receptor antagonist7 that features high receptor occupancy and full and long lasting (≥24hrs) central NK-1 receptor occupancy8. It has previously completed extensive safety and toxicology studies to support its clinical development; and it has already demonstrated a positive antidepressant effect in a Phase II clinical study together with beneficial effects on sleep8.
PATENT
NK1 antagonist compound orvepitant maleate, pharmaceutical formulations comprising this crystalline form, its use in therapy and processes for preparing the same. Background of the invention
WO03/066635 describes a number of diazabicycle derivatives having NK1 activity, including the 2-(R)-(4-Fluoro-2-methyl-phenyl)-4-(S)-((8aS)-6-oxo-hexahydro- pyrrolo[1 ,2-a]-pyrazin-2-yl)-piperidine-1-carboxylic acid [1-(R)-(3,5-bis-trifluoromethyl- phenyl)-ethyl]-methylamide (otherwise known as orvepitant).
The structure of the 2-(R)-(4-Fluoro-2-methyl-phenyl)-4-(S)-((8aS)-6-oxo-hexahydro- pyrrolo[1 ,2-a]-pyrazin-2-yl)-piperidine-1-carboxylic acid [1-(R)-(3,5-bis-trifluoromethyl- phenyl)-ethyl]-methylamide (otherwise known as orvepitant) is shown in formula (I) below:
 Hereinafter any reference to orvepitant refers to the compound of formula (I).
Hereinafter any reference to orvepitant refers to the compound of formula (I).
Orvepitant may also be known as: CAS Index name
1-Piperidinecarboxamide, Λ/-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-2-(4-fluoro-
2-methylphenyl)-4-[(8aS)-hexahydro-6-oxopyrrolo[1 ,2-a]pyrazin-2(1 /-/)-yl]-Λ/-methyl-,
(2RAS) and IUPAC name :
(2R,4S)-Λ/-{(1 R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-
Λ/-methyl-4-[(8aS)-6-oxohexahydropyrrolo[1 ,2-a]pyrazin-2(1 /-/)-yl]-1- piperidinecarboxamide. A preferred salt of this compound is its hydrochloride salt which is otherwise known as orvepitant hydrochloride.
A further preferred salt of this compound is its maleate salt which is otherwise known as orvepitant maleate.
Particularly Example 1 1 C of WO03/066635 describes the synthesis of orvepitant maleate using substantially the same experimental conditions described in the Example 1 in the present patent application.
We have now found that orvepitant maleate can be obtained in a new crystalline form. In particular, we have discovered a form of orvepitant maleate which is anhydrous and crystalline and which surprisingly has particularly good pharmaceutical properties. This is particularly stable and essentially non hygroscopic. It also has good storage properties and can be readily formulated into pharmaceutical compositions such as tablets and capsules.
Example 1 : preparation of orvepitant maleate (Form 2) {(1 R)-1 -[3,5-bis(trifluoromethyl)phenyl]ethyl}methylamine - (2R)-2-hydroxybutanedioic acid (1.8 kg) was added to ethyl acetate (5.4 litres) and 15% w/w sodium carbonate solution (5.4 litres) and was stirred until all solids had dissolved. The organic phase was separated and was washed with water (5.4 litres). Fresh ethyl acetate (6.7 litres) was added and the solution was distilled to 5.4 litres under reduced pressure.
The solution was diluted with ethyl acetate (3.6 litres). The reactor was purged with carbon dioxide and a continuous steady stream of carbon dioxide was maintained. Triethylamine (810 ml) was added over 30 minutes and was rinsed in with ethyl acetate (250 ml). The reaction mixture was stirred for 30 minutes. Chlorotrimethylsilane (850 ml) was added over 30 minutes with cooling to keep the temperature between 17°C and 23°C and was rinsed in with ethyl acetate (250 ml). The reaction mixture was stirred for 30 minutes. Pyridine (720 ml) was added and was rinsed in with ethyl acetate (250 ml). Thionyl chloride (480 ml) was added over 10 minutes and then a rinse of ethyl acetate (500 ml). The reaction mixture was stirred at 200C for 16 hours under a carbon dioxide atmosphere.
28% w/w Racemic malic acid solution (5.3 litres) was added and the mixture was stirred for 15 minutes. The organic phase was separated, diluted with ethyl acetate (1.5 litres) and was washed with water (2 x 2.7 litres) and 20% w/w dibasic potassium phosphate solution (5.6 litres). The solution was distilled under reduced pressure to a total volume of 2.5 litres. Ethyl acetate (5 litres) was added and the solution was redistilled to 3 litres to give a solution of {(1 R)-1-[3,5- bis(trifluoromethyl)phenyl]ethyl}methylcarbamic chloride.
(2R)-2-(4-fluoro-2-methylphenyl)-4-piperidinone - (2S)-hydroxy(phenyl)ethanoic acid (1.2 kg) was added to 15% w/w sodium carbonate solution (4.8 litres) and ethyl acetate (4.8 litres) and the mixture was stirred until solids dissolved. The organic phase was separated and was washed with 20% w/w sodium chloride solution (4 litres). Fresh ethyl acetate (4.8 litres) was added and the solution of (2R)-2-(4-fluoro- 2-methylphenyl)-4-piperidinone was distilled under reduced pressure to a volume of 3 litres. The solution of (2R)-2-(4-fluoro-2-methylphenyl)-4-piperidinone was charged to the solution of {(1 R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl}methylcarbamic chloride followed by an ethyl acetate (300 ml) rinse. Triethylamine (857 g) was added followed by ethyl acetate (300 ml) and the mixture was boiled at reflux for 18 hours. The slurry was cooled to 200C and N-acetylpiperazine (240 g) was added. The reaction mixture was stirred for 30 minutes at 200C and was then charged with 28% w/w racemic malic acid solution (3.6 litres). The organic phase was separated and was washed with 20% w/w sodium chloride solution (4.8 litres). Ethyl acetate (4.8 litres) was added and the solution of (2R)-N-{(1 R)-1-[3,5- bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-4-oxo-1- piperidinecarboxamide was distilled under reduced pressure distillation to a total volume of 3 litres.
(8aS)-hexahydropyrrolo[1 ,2-a]pyrazin-6(2H)-one - (2S)-(acetyloxy)(phenyl)ethanoic acid (1.5 kg) was added to acetonitrile (11.4 litres) and triethylamine (450 g) was added. An acetonitrile (250 ml) rinse was added and the slurry was stirred at 200C for 30 min. Sodium triacetoxyborohydride (900 g) was added and the reaction was cooled to 100C. Formic acid (396 ml) was added to the mixture over 30 min, maintaining the temperature below 15°C. An acetonitrile (250 ml) rinse was added and the reaction was warmed to 200C. The solution of (2R)-N-{(1 R)-1-[3,5- bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-4-oxo-1- piperidinecarboxamide in ethyl acetate was added to the reaction mixture and was rinsed in with acetonitrile (1 litre). The reaction was stirred for 16 hours at 200C.
The slurry was distilled to 5 litres under reduced pressure. The mixture was diluted with ethyl acetate (10 litres) and was washed with 13% w/w ammonia solution (2 x 4 litres), and 10% w/w sodium chloride solution (4 litres). The organic solution was distilled to 5 litres under reduced pressure. The solution was diluted with IPA (8 litres) and was distilled under reduced pressure to 5 litres. Further IPA (8 litres) was added and the solution was again distilled to 5 litres.
A solution of maleic acid (248.5 g) in IPA (2.5 litres) was added. The mixture was then seeded with orvepitant maleate A (1 g) and the mixture was aged for 1 hour. Iso-octane (10 litres) was added over 30 min. and the mixture further aged for 1 hour. The slurry was cooled to 7°C and was further aged for 90 minutes. The solid formed was filtered and washed with a 1 :1 mixture of IPA/iso-octane (2 x 3 litres). The resulting solid was dried at 40°C under reduced pressure to give the title compound (1.095kg, 44%). NMR (CD3OD) δ (ppm) 1.52-1.53 (d, 3H), 1.68-1.78 (m, 1 H), 1.82-1.91 (q, 1 H), 1.95- 2.05 (m, 1 H), 2.16-2.37 (m, 3H), 2.38-2.50 (m, 2H), 2.44 (s, 3H), 2.81-2.87 (t, 1 H),
2.83 (s, 3H), 2.90-2.99 (m, 2H), 3.1 1-3.18 (dt, 1 H), 3.48-3.60 (m, 3H), 3.66-3.69 (d, 1 H), 3.89-3.96 (m, 1 H), 4.15-4.19 (dd, 1 H), 4.33-4.36 (dd , 1 H), 5.40-5.45 (q, 1 H), 6.26 (s, 2H), 6.76-6.81 (dt, 1 H), 6.85-6.88 (dd, 1 H), 7.27-7.31 (dd, 1 H), 7.70 (s, 2H), 7.88 (s, 1 H). (M+H)+ Calcd for C3iH35F7N4O 629, found 629.
References:
WO03/066635 describes a number of diazabicycle derivatives having NK1 activity, including the 2-(R)-(4-Fluoro-2-methyl-phenyl)-4-(S)-((8aS)-6-oxo-hexahydro- pyrrolo[1 ,2-a]-pyrazin-2-yl)-piperidine-1-carboxylic acid [1-(R)-(3,5-bis-trifluoromethyl- phenyl)-ethyl]-methylamide (otherwise known as orvepitant).
The structure of the 2-(R)-(4-Fluoro-2-methyl-phenyl)-4-(S)-((8aS)-6-oxo-hexahydro- pyrrolo[1 ,2-a]-pyrazin-2-yl)-piperidine-1-carboxylic acid [1-(R)-(3,5-bis-trifluoromethyl- phenyl)-ethyl]-methylamide (otherwise known as orvepitant) is shown in formula (I) below:

Orvepitant may also be known as: CAS Index name
1-Piperidinecarboxamide, Λ/-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-2-(4-fluoro-
2-methylphenyl)-4-[(8aS)-hexahydro-6-oxopyrrolo[1 ,2-a]pyrazin-2(1 /-/)-yl]-Λ/-methyl-,
(2RAS) and IUPAC name :
(2R,4S)-Λ/-{(1 R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-
Λ/-methyl-4-[(8aS)-6-oxohexahydropyrrolo[1 ,2-a]pyrazin-2(1 /-/)-yl]-1- piperidinecarboxamide. A preferred salt of this compound is its hydrochloride salt which is otherwise known as orvepitant hydrochloride.
A further preferred salt of this compound is its maleate salt which is otherwise known as orvepitant maleate.
Particularly Example 1 1 C of WO03/066635 describes the synthesis of orvepitant maleate using substantially the same experimental conditions described in the Example 1 in the present patent application.
We have now found that orvepitant maleate can be obtained in a new crystalline form. In particular, we have discovered a form of orvepitant maleate which is anhydrous and crystalline and which surprisingly has particularly good pharmaceutical properties. This is particularly stable and essentially non hygroscopic. It also has good storage properties and can be readily formulated into pharmaceutical compositions such as tablets and capsules.
Example 1 : preparation of orvepitant maleate (Form 2) {(1 R)-1 -[3,5-bis(trifluoromethyl)phenyl]ethyl}methylamine - (2R)-2-hydroxybutanedioic acid (1.8 kg) was added to ethyl acetate (5.4 litres) and 15% w/w sodium carbonate solution (5.4 litres) and was stirred until all solids had dissolved. The organic phase was separated and was washed with water (5.4 litres). Fresh ethyl acetate (6.7 litres) was added and the solution was distilled to 5.4 litres under reduced pressure.
The solution was diluted with ethyl acetate (3.6 litres). The reactor was purged with carbon dioxide and a continuous steady stream of carbon dioxide was maintained. Triethylamine (810 ml) was added over 30 minutes and was rinsed in with ethyl acetate (250 ml). The reaction mixture was stirred for 30 minutes. Chlorotrimethylsilane (850 ml) was added over 30 minutes with cooling to keep the temperature between 17°C and 23°C and was rinsed in with ethyl acetate (250 ml). The reaction mixture was stirred for 30 minutes. Pyridine (720 ml) was added and was rinsed in with ethyl acetate (250 ml). Thionyl chloride (480 ml) was added over 10 minutes and then a rinse of ethyl acetate (500 ml). The reaction mixture was stirred at 200C for 16 hours under a carbon dioxide atmosphere.
28% w/w Racemic malic acid solution (5.3 litres) was added and the mixture was stirred for 15 minutes. The organic phase was separated, diluted with ethyl acetate (1.5 litres) and was washed with water (2 x 2.7 litres) and 20% w/w dibasic potassium phosphate solution (5.6 litres). The solution was distilled under reduced pressure to a total volume of 2.5 litres. Ethyl acetate (5 litres) was added and the solution was redistilled to 3 litres to give a solution of {(1 R)-1-[3,5- bis(trifluoromethyl)phenyl]ethyl}methylcarbamic chloride.
(2R)-2-(4-fluoro-2-methylphenyl)-4-piperidinone - (2S)-hydroxy(phenyl)ethanoic acid (1.2 kg) was added to 15% w/w sodium carbonate solution (4.8 litres) and ethyl acetate (4.8 litres) and the mixture was stirred until solids dissolved. The organic phase was separated and was washed with 20% w/w sodium chloride solution (4 litres). Fresh ethyl acetate (4.8 litres) was added and the solution of (2R)-2-(4-fluoro- 2-methylphenyl)-4-piperidinone was distilled under reduced pressure to a volume of 3 litres. The solution of (2R)-2-(4-fluoro-2-methylphenyl)-4-piperidinone was charged to the solution of {(1 R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl}methylcarbamic chloride followed by an ethyl acetate (300 ml) rinse. Triethylamine (857 g) was added followed by ethyl acetate (300 ml) and the mixture was boiled at reflux for 18 hours. The slurry was cooled to 200C and N-acetylpiperazine (240 g) was added. The reaction mixture was stirred for 30 minutes at 200C and was then charged with 28% w/w racemic malic acid solution (3.6 litres). The organic phase was separated and was washed with 20% w/w sodium chloride solution (4.8 litres). Ethyl acetate (4.8 litres) was added and the solution of (2R)-N-{(1 R)-1-[3,5- bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-4-oxo-1- piperidinecarboxamide was distilled under reduced pressure distillation to a total volume of 3 litres.
(8aS)-hexahydropyrrolo[1 ,2-a]pyrazin-6(2H)-one - (2S)-(acetyloxy)(phenyl)ethanoic acid (1.5 kg) was added to acetonitrile (11.4 litres) and triethylamine (450 g) was added. An acetonitrile (250 ml) rinse was added and the slurry was stirred at 200C for 30 min. Sodium triacetoxyborohydride (900 g) was added and the reaction was cooled to 100C. Formic acid (396 ml) was added to the mixture over 30 min, maintaining the temperature below 15°C. An acetonitrile (250 ml) rinse was added and the reaction was warmed to 200C. The solution of (2R)-N-{(1 R)-1-[3,5- bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-4-oxo-1- piperidinecarboxamide in ethyl acetate was added to the reaction mixture and was rinsed in with acetonitrile (1 litre). The reaction was stirred for 16 hours at 200C.
The slurry was distilled to 5 litres under reduced pressure. The mixture was diluted with ethyl acetate (10 litres) and was washed with 13% w/w ammonia solution (2 x 4 litres), and 10% w/w sodium chloride solution (4 litres). The organic solution was distilled to 5 litres under reduced pressure. The solution was diluted with IPA (8 litres) and was distilled under reduced pressure to 5 litres. Further IPA (8 litres) was added and the solution was again distilled to 5 litres.
A solution of maleic acid (248.5 g) in IPA (2.5 litres) was added. The mixture was then seeded with orvepitant maleate A (1 g) and the mixture was aged for 1 hour. Iso-octane (10 litres) was added over 30 min. and the mixture further aged for 1 hour. The slurry was cooled to 7°C and was further aged for 90 minutes. The solid formed was filtered and washed with a 1 :1 mixture of IPA/iso-octane (2 x 3 litres). The resulting solid was dried at 40°C under reduced pressure to give the title compound (1.095kg, 44%). NMR (CD3OD) δ (ppm) 1.52-1.53 (d, 3H), 1.68-1.78 (m, 1 H), 1.82-1.91 (q, 1 H), 1.95- 2.05 (m, 1 H), 2.16-2.37 (m, 3H), 2.38-2.50 (m, 2H), 2.44 (s, 3H), 2.81-2.87 (t, 1 H),
2.83 (s, 3H), 2.90-2.99 (m, 2H), 3.1 1-3.18 (dt, 1 H), 3.48-3.60 (m, 3H), 3.66-3.69 (d, 1 H), 3.89-3.96 (m, 1 H), 4.15-4.19 (dd, 1 H), 4.33-4.36 (dd , 1 H), 5.40-5.45 (q, 1 H), 6.26 (s, 2H), 6.76-6.81 (dt, 1 H), 6.85-6.88 (dd, 1 H), 7.27-7.31 (dd, 1 H), 7.70 (s, 2H), 7.88 (s, 1 H). (M+H)+ Calcd for C3iH35F7N4O 629, found 629.
References:
- Rosen AC et al. Am J Clin Dermatol. (2013), 14(4):327-33
- Santini D et al. Lancet Oncol. (2012), 13(10):1020-4
- Ensslin CJ et al. J Am Acad Dermatol. (2013), 69(5):708-20
- Duval A, Dubertret L. N Engl J Med. (2009), 1;361(14):1415-6
- Ständer S et al. PLoS One. (2010), 5(6):e10968
- Torres T et al. J Am Acad Dermatol. (2012), 66(1):e14-5
- Di Fabio R et al. Bioorg Med Chem. (2013), 21(21):6264-73
- Ratti E et al. J Psychopharmacol. (2013), 27(5):424-34
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015238486 | 2015-08-27 | NOVEL USES |
| US2014128395 | 2014-05-08 | Novel Method |
| US2011166150 | 2011-07-07 | Anhydrous Crystal Form Of Ovrepitant Maleate |
| US2010317666 | 2010-12-16 | Composition Comprising An NK-1 Receptor Antagonist And An SSRI For The Treatment Of Tinnitus And Hearing Loss |
| US2010152446 | 2010-06-17 | Piperidine Derivatives |
| US2010105688 | 2010-04-29 | PHARMACEUTICAL COMPOSITIONS COMPRISING 3,5-DIAMINO-6-(2,3-DICHLOPHENYL)-1,2,4-TRIAZINE OR R(-)-2,4-DIAMINO-5-(2,3-DICHLOROPHENYL)-6-FLUOROMETHYL PYRIMIDINE AND AN NK1 |
| US7652012 | 2010-01-26 | 2-(R)-(4-fluoro-2-methyl-phenyl)-4-(S)-((8aS)-6-oxo-hexahydro-pyrrolo[1,2-a]-pyrazin-2-yl)-piperidine-1-carboxylic acid [1-(R)-3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamide maleate and pharmaceutical compositions thereof |
| US2009326032 | 2009-12-31 | PHARMACEUTICAL COMPOSITIONS COMPRISING NK1 RECEPTOR ANTAGONISTS AND SODIUM CHANNEL BLOCKERS |
| US2009318530 | 2009-12-24 | PHARMACEUTICAL COMPOSITIONS COMPRISING NK1 RECEPTOR ANTAGONISTS AND SODIUM CHANNEL BLOCKERS |
| US7189713 | 2007-03-13 | Piperidine derivatives |
| Patent ID | Date | Patent Title |
|---|---|---|
| US7189713 | 2007-03-13 | Piperidine derivatives |
| US2006287325 | 2006-12-21 | Combinations of paroxetine and 2-(r)-(4-fluoro-2-methyl-phenyl)-4-(s)-((8as)-6-oxo-hexahydro-pyrrolo'1,2-a!-pyrazin-2-yl)-piperidine-1-carboxylicacid'1-(r)-(3,5-bis-trifluoromethyl-phenyl)- |
| US6384099 | 2002-05-07 | Method for curing polymeric materials, such as those used in dentistry, and for tailoring the post-cure properties of polymeric materials through the use of light source power modulation |
| US6282013 | 2001-08-28 | System for curing polymeric materials, such as those used in dentistry, and for tailoring the post-cure properties of polymeric materials through the use of light source power modulation |
| US6008264 | 1999-12-28 | Method for curing polymeric materials, such as those used in dentistry, and for tailoring the post-cure properties of polymeric materials through the use of light source power modulation |
REFERENCES
1: Di Fabio R, Alvaro G, Braggio S, Carletti R, Gerrard PA, Griffante C, Marchioro C, Pozzan A, Melotto S, Poffe A, Piccoli L, Ratti E, Tranquillini E, Trower M, Spada S, Corsi M. Identification, biological characterization and pharmacophoric analysis of a new potent and selective NK1 receptor antagonist clinical candidate. Bioorg Med Chem. 2013 Nov 1;21(21):6264-73. doi: 10.1016/j.bmc.2013.09.001. Epub 2013 Sep 11. PubMed PMID: 24075145.
2: Ratti E, Bettica P, Alexander R, Archer G, Carpenter D, Evoniuk G, Gomeni R, Lawson E, Lopez M, Millns H, Rabiner EA, Trist D, Trower M, Zamuner S, Krishnan R, Fava M. Full central neurokinin-1 receptor blockade is required for efficacy in depression: evidence from orvepitant clinical studies. J Psychopharmacol. 2013 May;27(5):424-34. doi: 10.1177/0269881113480990. Epub 2013 Mar 28. PubMed PMID: 23539641.
///////Orvepitant, GW823296, PHASE 2, Neurokinin 1 (NK1) receptor antagonist
C[C@@H](N(C)C(=O)N1CC[C@@H](C[C@@H]1c1ccc(F)cc1C)N1CCN2[C@@H](CCC2=O)C1)c1cc(cc(c1)C(F)(F)F)C(F)(F)F
CC1=C(C=CC(=C1)F)C2CC(CCN2C(=O)N(C)C(C)C3=CC(=CC(=C3)C(F)(F)F)C(F)(F)F)N4CCN5C(C4)CCC5=O