FLOZIN SERIES 2/2

FULL LENGTH ARTICLES
1 TOFOGLIFLOZIN
2 SERGLIFLOZIN
3 DAPAGLIFLOZIN
4 IPRAGLIFLOZIN
5 EMPAGLIFLOZIN
6 LUSEOGLIFLOZIN
7 REMOGLIFLOZIN
8 ERTUGLIFLOZIN
9 SOTAGLIFLOZON
10 CANAGLIFLOZIN
SEE PART 1 AT .......http://medcheminternational.blogspot.in/p/flozin-series.html
SEE PART 1 AT .......http://medcheminternational.blogspot.in/p/flozin-series.html
PART 2
11 SBM-TFC-039
12 LIK 066
13 BEXAGLIFLOZIN
14
15
will be Updated with atigliflozin etc
11
SBM-TFC-039
SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem !!
A new “flozin” seems to me appearing on the horizon in form of SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem, picked up a list from WO 2012160218, from TFChem…….see link , Sirona Biochem Announces SGLT2 Inhibitor and Skin Lightening Patent Granted, 29 Jun 2015, Patent entitled “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
This led me to search, “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
WO 2012160218 A1, IN 2013-DN10635, CN 103649033Tf化学公司
| Applicant | Tfchem |

List above as in http://www.google.com/patents/WO2012160218A1?cl=en
FROM THE ABOVE LIST, SBM-TFC-039 MAY BE PREDICTED/OR AS SHOWN BELOW
COMPD 16 as in/WO2012160218
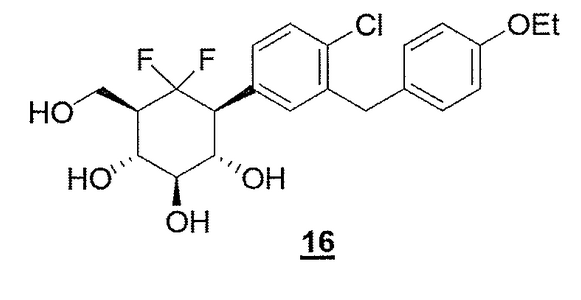
COMPD 16, PREDICTED/LIKELY SBM-TFC-039 has CAS 1413373-30-4, name D-myo-Inositol, 1-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1,2,3-trideoxy-2,2-difluoro-3-(hydroxymethyl)-
Just scrolling through the patent gave me more insight
MORE EVIDENCE….http://www.google.com/patents/WO2012160218A1?cl=en, this patent descibes compd 16 as follows
Compound 16 according to the invention has been compared to Dapaglifozin to underline the improvement of the duration of action, i.e. the longer duration of glucosuria, of the compound when the intracyclic oxygen atom of the glucose moiety is replaced by a CF2 moiety.

The results obtained are presented on Figure 5. It appears thus that 16 (3 mg/kg) triggered glucosuria that lasted beyond 24 hours compared to Dapagliflozin.
• Compound 16 according to the invention has been compared to the compound 9 of WO 2009/1076550 to underline the improvement of the duration of action of the compound when a mimic of glucose bearing a CH-OH moiety instead of the intracyclic oxygen atom is replaced by a mimic of glucose bearing a CF2 in place of the CH-OH moiet .

NOTE=COMPD 9 OF WO 2009/1076550 has CAS 1161430-16-5, D-scyllo-
Inositol,
1-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1,3-dideoxy-3-
(hydroxymethyl)- and is very similar to the compd under discussion

| Company | Sirona Biochem Corp. |
| Description | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Molecular Target | Sodium-glucose cotransporter 2 (SGLT2) |
| Mechanism of Action | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
| Regulatory Designation | |
| Partner | Shanghai Fosun Pharmaceutical Group Co. Ltd. |
SBM-TFC-039
PATENT
WO 2012160218
http://www.google.com/patents/WO2012160218A1?cl=en
Examples within this first subclass include but are not limited to:

Synthesis of compound 8
C35H34O5 M = 534.64 g.mol“
Mass: (ESI ): 535.00 (M + H); 552.00 (M + H20); 785.87; 1086.67 (2M + H20)
 A.
A.
 Procedure A:
Procedure A:
To a solution of 4 (10.5g, 15.89mmol, leq) in toluene (400mL) were added 18-crown-6 (168mg, 0.64mmol, 0.04eq) and potassium carbonate (6.69g, 48.5mmol, 3.05eq.). The mixture was stirred overnight at room temperature, and then the remising insoluble material was filtered off and washed with toluene. The filtrate and the washings were combined, washed with 2N hydrochloric acid aqueous solution followed by saturated sodium hydrogencarbonate aqueous solution, dried over sodium sulphate, filtered and concentrated under reduced pressure. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford cyclohexenone 8 (4.07g; 48% yield) as yellowish oil.
Procedure B:
A solution of 7 (3.27g, 5.92mmol, leq) in pyridine (14mL) was cooled to 0°C before POCl3 (2.75mL, 29.6mmol, 5eq) was added dropwise. The mixture was stirred at this temperature for 10 min before the cooling bath was removed. The reaction mixture was stirred overnight at room temperature before being re-cooled to 0°C. POCI3 (2.75mL, 29.6mmol, 5eq) was added once again trying to complete the reaction. The mixture was stirred for an additional 20h at room temperature before being diluted with Et20 (20mL) and poured onto crushed ice. 1M HC1 aqueous solution (lOOmL) was added, and the mixture was extracted with Et20 (200mL & l OOmL). The combined organic extracts were washed with brine (lOOmL), dried over sodium sulphate, filtered and concentrated before being purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 80:20) to afford compound 8 (1.46g, 46% yield) as an orange oil. Synthesis of compound 9
C15H12BrC102 M = 339.61 g.moF1
Mass: (GC-MS): 338-340
 The synthesis of this product is described in J. Med. Chem. 2008, 51, 1 145—1149.Synthesis of compound 10
The synthesis of this product is described in J. Med. Chem. 2008, 51, 1 145—1149.Synthesis of compound 10
C15H14B1CIO M = 325.63 g.mof1
 10 The synthesis of this product is described in J. Med. Chem. 2008, 51, 1145-1 149.
10 The synthesis of this product is described in J. Med. Chem. 2008, 51, 1145-1 149.
Synthesis of compound 11
C50H49CIO6 M = 781.37 g.moF1
Mass: ESI+): 798.20 (M + H20)
 Under inert atmosphere, Mg powder (265mg, 10.9mmol, 2.4eq) was
charged into a three necked flask, followed by addition of a portion of
1/3 of a solution of the 4- bromo-l-chloro-2-(4-ethylbenzyl)benzene
(2.95g, 9.1mmol; 2eq) in dry THF (25mL) and 1 ,2-dibromoethane (10 mol %
of Mg; 85mg; 0.45mmol). The mixture was heated to reflux. After the
reaction was initiated (exothermic and consuming of Mg), the remaining
solution of 2-(4-ethylbenzyl)-4-bromo-l-chlorobenzene in dry TFIF was
added dropwise. The mixture was then allowed to react for another one
hour under gentle reflux until most of the Mg was consumed.
Under inert atmosphere, Mg powder (265mg, 10.9mmol, 2.4eq) was
charged into a three necked flask, followed by addition of a portion of
1/3 of a solution of the 4- bromo-l-chloro-2-(4-ethylbenzyl)benzene
(2.95g, 9.1mmol; 2eq) in dry THF (25mL) and 1 ,2-dibromoethane (10 mol %
of Mg; 85mg; 0.45mmol). The mixture was heated to reflux. After the
reaction was initiated (exothermic and consuming of Mg), the remaining
solution of 2-(4-ethylbenzyl)-4-bromo-l-chlorobenzene in dry TFIF was
added dropwise. The mixture was then allowed to react for another one
hour under gentle reflux until most of the Mg was consumed.
The above Grignard reagent was added dropwise into the solution of cyclohexenone 8 (2.42g, 4.53mmol, leq) in dry THF (25mL) under inert atmosphere at room temperature (about 25°C), then allowed to react for 3h. A saturated aqueous solution of ammonium chloride was added into the mixture to quench the reaction. The mixture was extracted with Et20, washed with brine, dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 100:0 to 80:20) to afford the target compound 11 as a yellow oil (3.01g, 86%).
Synthesis of compound 12
C5oH49C105 M = 765.37 g.mol“1
+): 782.13 (M + H20)
 Triethylsilane (0.210mL, 1.30mmol, 3eq) and boron-trifluoride etherate (48% BF3,
O. l lOmL, 0.866mmol, 2eq) were successively added into a solution of
alcohol 1 1 (338mg, 0.433mmol, leq) in dichloromethane (5mL) under inert
atmosphere at -20°C. After stirring for 2.5h, a saturated aqueous
solution of sodium chloride was added to quench the reaction. The
mixture was extracted with CH2C12 (10mLx3) and the organic layer was washed with brine, dried over Na2S04,
filtrated and concentrated. The residue was purified on silica gel
chromatography (cyclohexane/ethyl acetate 9.8:0.2 to 8:2) to afford the
target compound 12 as a white powder (278 mg, 0.363mmol, 84%).
Triethylsilane (0.210mL, 1.30mmol, 3eq) and boron-trifluoride etherate (48% BF3,
O. l lOmL, 0.866mmol, 2eq) were successively added into a solution of
alcohol 1 1 (338mg, 0.433mmol, leq) in dichloromethane (5mL) under inert
atmosphere at -20°C. After stirring for 2.5h, a saturated aqueous
solution of sodium chloride was added to quench the reaction. The
mixture was extracted with CH2C12 (10mLx3) and the organic layer was washed with brine, dried over Na2S04,
filtrated and concentrated. The residue was purified on silica gel
chromatography (cyclohexane/ethyl acetate 9.8:0.2 to 8:2) to afford the
target compound 12 as a white powder (278 mg, 0.363mmol, 84%).
Synthesis of compound 13
C5oH5tC106 M = 783.39g.moF1
Mass: (ESI+): 800 (M + H20); 1581 (2M + H20)
 Under inert atmosphere, borane-dimethyl sulfide complex (2M in THF,
16.7mL, 33mmol, 10.5eq) was added to a solution of 12 (2.41g; 3.15mmol,
leq) in dry THF (lOOmL) cooled to 0°C. The reaction mixture was then
refluxed for lh,cooled to 0°C and treated carefully with sodium
hydroxide (3M in H20, 10.5mL, 31.5mmol, lOeq), followed by hydrogen peroxide (30% in H20,
3.2mL, 31.5mmol, l Oeq) at room temperature (above 30°C). The mixture
was allowed to react overnight at room temperature (~25°C) before a
saturated aqueous solution of ammonium chloride was added to quench the
reaction. The mixture was extracted with ethyl acetate and the organic
layer was washed with brine, dried over Na2S04,
filtered, and concentrated. The residue was purified by silica gel
chromatography (cyclohexane/ethyl acetate 97:3 to 73:27) to afford the
desired compound 13 (1.05g; 43%) as a yellowish oil.
Under inert atmosphere, borane-dimethyl sulfide complex (2M in THF,
16.7mL, 33mmol, 10.5eq) was added to a solution of 12 (2.41g; 3.15mmol,
leq) in dry THF (lOOmL) cooled to 0°C. The reaction mixture was then
refluxed for lh,cooled to 0°C and treated carefully with sodium
hydroxide (3M in H20, 10.5mL, 31.5mmol, lOeq), followed by hydrogen peroxide (30% in H20,
3.2mL, 31.5mmol, l Oeq) at room temperature (above 30°C). The mixture
was allowed to react overnight at room temperature (~25°C) before a
saturated aqueous solution of ammonium chloride was added to quench the
reaction. The mixture was extracted with ethyl acetate and the organic
layer was washed with brine, dried over Na2S04,
filtered, and concentrated. The residue was purified by silica gel
chromatography (cyclohexane/ethyl acetate 97:3 to 73:27) to afford the
desired compound 13 (1.05g; 43%) as a yellowish oil.
Synthesis of compound 14
C50H49CIO6 M = 781.37g.mol“1
Mass: (ESI+): 798 (M + H20); 1471; 1579 (2M + H20)
 13 14
13 14
Dess-Martin periodinane (81mg; 1.91mmol; 1.5eq) was added portion wise to a solution of alcohol 13 (l .Og; 1.28mmol, leq) in anhydrous dichloromethane (20mL) at 0°C. The reaction was then stirred overnight at room temperature before being quenched with IN aqueous solution of sodium hydroxide. The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 82: 18), to afford the target ketone 14 (783mg, 79% yield) as a colorless oil. Synthesis of compound 15
C5oH49ClF206 M = 803.37g.moF1
19 F NMR (CDCU, 282.5MHz): -100.3 (d, J=254Hz, IF, CFF); -1 13.3 (td, Jl=254Hz, J2=29Hz, IF, CFF).
Mass: (ESI+): 820.00 (M+H20)
 14 15
14 15
A solution of ketone 14 (421mg, 0.539mmol, leq) in DAST (2mL, 16.3mmol, 30eq.) was stirred under inert atmosphere at 70°C for 12h. The mixture was then cooled to room temperature and dichloromethane was added. The solution was poured on a mixture of water, ice and solid NaHC03. Agitation was maintained for 30min while reaching room temperature. The aqueous layer was extracted with dichloromethane and the organic phase was dried over Na2S04, filtered and concentrated. The crude product was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford the desired compound 15 as a yellowish oil ( 182mg, 42% yield).
Synthesis of compound 16
C22H25CIF2O5 M = 442.88g.mor1
19 F NMR (MeOD, 282.5MHz): -96.7 (d, J=254Hz, IF, CFF); 12.2 (td,
Jl=254Hz, J2=28Hz, IF, CFF).
Mass: (ESI+): 465.3 (M+Na)
 o-Dichlorobenzene (0.320mL, 2.82mol, lOeq) followed by Pd/C 10%
(0.342g, 0.32mol, l .leq) were added to a solution of 15 (228mg,
0.28mmol, leq) in a mixture of THF and MeOH (2: 1, v/v, 160mL). The
reaction was placed under hydrogen atmosphere and stirred at room
temperature for 2h. The reaction mixture was filtered and concentrated
before being purified on silica gel chromatography
(dichloromethane/methanol 100: 1 to 90: 10) to afford compound 16
(105mg, 83% yield).
o-Dichlorobenzene (0.320mL, 2.82mol, lOeq) followed by Pd/C 10%
(0.342g, 0.32mol, l .leq) were added to a solution of 15 (228mg,
0.28mmol, leq) in a mixture of THF and MeOH (2: 1, v/v, 160mL). The
reaction was placed under hydrogen atmosphere and stirred at room
temperature for 2h. The reaction mixture was filtered and concentrated
before being purified on silica gel chromatography
(dichloromethane/methanol 100: 1 to 90: 10) to afford compound 16
(105mg, 83% yield).
C35H34O5 M = 534.64 g.mol“
Mass: (ESI ): 535.00 (M + H); 552.00 (M + H20); 785.87; 1086.67 (2M + H20)


To a solution of 4 (10.5g, 15.89mmol, leq) in toluene (400mL) were added 18-crown-6 (168mg, 0.64mmol, 0.04eq) and potassium carbonate (6.69g, 48.5mmol, 3.05eq.). The mixture was stirred overnight at room temperature, and then the remising insoluble material was filtered off and washed with toluene. The filtrate and the washings were combined, washed with 2N hydrochloric acid aqueous solution followed by saturated sodium hydrogencarbonate aqueous solution, dried over sodium sulphate, filtered and concentrated under reduced pressure. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford cyclohexenone 8 (4.07g; 48% yield) as yellowish oil.
Procedure B:
A solution of 7 (3.27g, 5.92mmol, leq) in pyridine (14mL) was cooled to 0°C before POCl3 (2.75mL, 29.6mmol, 5eq) was added dropwise. The mixture was stirred at this temperature for 10 min before the cooling bath was removed. The reaction mixture was stirred overnight at room temperature before being re-cooled to 0°C. POCI3 (2.75mL, 29.6mmol, 5eq) was added once again trying to complete the reaction. The mixture was stirred for an additional 20h at room temperature before being diluted with Et20 (20mL) and poured onto crushed ice. 1M HC1 aqueous solution (lOOmL) was added, and the mixture was extracted with Et20 (200mL & l OOmL). The combined organic extracts were washed with brine (lOOmL), dried over sodium sulphate, filtered and concentrated before being purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 80:20) to afford compound 8 (1.46g, 46% yield) as an orange oil. Synthesis of compound 9
C15H12BrC102 M = 339.61 g.moF1
Mass: (GC-MS): 338-340

C15H14B1CIO M = 325.63 g.mof1

Synthesis of compound 11
C50H49CIO6 M = 781.37 g.moF1
Mass: ESI+): 798.20 (M + H20)

The above Grignard reagent was added dropwise into the solution of cyclohexenone 8 (2.42g, 4.53mmol, leq) in dry THF (25mL) under inert atmosphere at room temperature (about 25°C), then allowed to react for 3h. A saturated aqueous solution of ammonium chloride was added into the mixture to quench the reaction. The mixture was extracted with Et20, washed with brine, dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 100:0 to 80:20) to afford the target compound 11 as a yellow oil (3.01g, 86%).
Synthesis of compound 12
C5oH49C105 M = 765.37 g.mol“1
+): 782.13 (M + H20)

Synthesis of compound 13
C5oH5tC106 M = 783.39g.moF1
Mass: (ESI+): 800 (M + H20); 1581 (2M + H20)

Synthesis of compound 14
C50H49CIO6 M = 781.37g.mol“1
Mass: (ESI+): 798 (M + H20); 1471; 1579 (2M + H20)

Dess-Martin periodinane (81mg; 1.91mmol; 1.5eq) was added portion wise to a solution of alcohol 13 (l .Og; 1.28mmol, leq) in anhydrous dichloromethane (20mL) at 0°C. The reaction was then stirred overnight at room temperature before being quenched with IN aqueous solution of sodium hydroxide. The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 82: 18), to afford the target ketone 14 (783mg, 79% yield) as a colorless oil. Synthesis of compound 15
C5oH49ClF206 M = 803.37g.moF1
19 F NMR (CDCU, 282.5MHz): -100.3 (d, J=254Hz, IF, CFF); -1 13.3 (td, Jl=254Hz, J2=29Hz, IF, CFF).
Mass: (ESI+): 820.00 (M+H20)

A solution of ketone 14 (421mg, 0.539mmol, leq) in DAST (2mL, 16.3mmol, 30eq.) was stirred under inert atmosphere at 70°C for 12h. The mixture was then cooled to room temperature and dichloromethane was added. The solution was poured on a mixture of water, ice and solid NaHC03. Agitation was maintained for 30min while reaching room temperature. The aqueous layer was extracted with dichloromethane and the organic phase was dried over Na2S04, filtered and concentrated. The crude product was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford the desired compound 15 as a yellowish oil ( 182mg, 42% yield).
Synthesis of compound 16
C22H25CIF2O5 M = 442.88g.mor1
19 F NMR (MeOD, 282.5MHz): -96.7 (d, J=254Hz, IF, CFF); 12.2 (td,
Jl=254Hz, J2=28Hz, IF, CFF).
Mass: (ESI+): 465.3 (M+Na)

…………………….
CN 103649033
Sirona Biochem’s SGLT Inhibitor Performs Better Than Johnson and Johnson’s SGLT Inhibitor, According to Study
Vancouver, British Columbia – December 7, 2012 – Sirona Biochem Corp. (TSX-V: SBM), announced its sodium glucose transporter (SGLT) inhibitor for Type 2 diabetes reduced blood glucose more effectively than Johnson and Johnson’s canagliflozin,
an advanced SGLT inhibitor being considered for market approval in
Europe and the U.S. Studies compared Sirona Biochem’s SGLT Inhibitor,
SBM-TFC-039, with canagliflozin and were conducted on Zucker Diabetic
Fatty (ZDF) rats.
In the study, SBM-TFC-039 significantly and rapidly reduced blood glucose levels at a dose of 1.0 mg/kg. Six (6) hours after administration, SBM-TFC-039 reduced blood glucose by 44% compared to canagliflozin at 26%. SBM-TFC-039 also had a longer duration of effect than canagliflozin. At 36 and 48 hours after treatment, SBM-TFC-039, at a dose of 1.0 mg/kg, was still effective at reducing blood glucose, whereas canagliflozin lost its effect after 36 hours. Studies were conducted at the Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ) by Principal Investigator Dr. Denis Richard, Research Chair on Obesity and Professor, Faculty of Medicine, Department of Anatomy & Physiology at Laval University.
“SGLT Inhibitors are a ground-breaking new treatment for Type 2 diabetes and these results demonstrate that SBM-TFC-039 will be a significant competitor for other SGLT Inhibitors,” said Neil Belenkie, Chief Executive Officer of Sirona Biochem. “The first SGLT Inhibitor,Forxiga™, was approved last month by the European Commission. We believe there is tremendous market potential worldwide for SGLT Inhibitors in the treatment of diabetes.”
SBM-TFC-039 is a sodium glucose transporter (SGLT) inhibitor. SGLT inhibitors are a new class of drug candidates for the treatment of diabetes. In the kidneys, SGLT inhibitors reduce the reabsorption of glucose into the bloodstream by eliminating excess glucose into the urine.
About Sirona Biochem Corp.
Sirona Biochem is a biotechnology company developing diabetes therapeutics, skin depigmenting and anti-aging agents for cosmetic use, biological ingredients and cancer vaccine antigens. The company utilizes a proprietary chemistry technique to improve pharmaceutical properties of carbohydrate-based molecules. For more information visit www.sironabiochem.com.

Laboratory – France
TFChem
Voie de l’innovation
Pharma Parc II
Chaussée du Vexin
27100 Val de Reuil
France
Phone:+33(0)2.32.09.01.16
Fax:+33(0)2.32.25.07.64

……………………………………………………………………………….In the study, SBM-TFC-039 significantly and rapidly reduced blood glucose levels at a dose of 1.0 mg/kg. Six (6) hours after administration, SBM-TFC-039 reduced blood glucose by 44% compared to canagliflozin at 26%. SBM-TFC-039 also had a longer duration of effect than canagliflozin. At 36 and 48 hours after treatment, SBM-TFC-039, at a dose of 1.0 mg/kg, was still effective at reducing blood glucose, whereas canagliflozin lost its effect after 36 hours. Studies were conducted at the Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ) by Principal Investigator Dr. Denis Richard, Research Chair on Obesity and Professor, Faculty of Medicine, Department of Anatomy & Physiology at Laval University.
“SGLT Inhibitors are a ground-breaking new treatment for Type 2 diabetes and these results demonstrate that SBM-TFC-039 will be a significant competitor for other SGLT Inhibitors,” said Neil Belenkie, Chief Executive Officer of Sirona Biochem. “The first SGLT Inhibitor,Forxiga™, was approved last month by the European Commission. We believe there is tremendous market potential worldwide for SGLT Inhibitors in the treatment of diabetes.”
SBM-TFC-039 is a sodium glucose transporter (SGLT) inhibitor. SGLT inhibitors are a new class of drug candidates for the treatment of diabetes. In the kidneys, SGLT inhibitors reduce the reabsorption of glucose into the bloodstream by eliminating excess glucose into the urine.
About Sirona Biochem Corp.
Sirona Biochem is a biotechnology company developing diabetes therapeutics, skin depigmenting and anti-aging agents for cosmetic use, biological ingredients and cancer vaccine antigens. The company utilizes a proprietary chemistry technique to improve pharmaceutical properties of carbohydrate-based molecules. For more information visit www.sironabiochem.com.
Laboratory – France
TFChem
Voie de l’innovation
Pharma Parc II
Chaussée du Vexin
27100 Val de Reuil
France
Phone:+33(0)2.32.09.01.16
Fax:+33(0)2.32.25.07.64
Shanghai Fosun Pharmaceutical Group Co. Ltd.

//////
12
LIK 066


LIK-066, a new flozin on the horizon
C23 H28 O7 . 2 C6 H11 N O, 642.7795, 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C…WO2011048112
CAS 1291095-45-8, (1S)-1,5-anhydro-1-C-[3- [(2,3-dihydro-1,4-benzodioxin-6-yl)methyl]-4-ethylphenyl]- D-glucitol (1:1) WITH L-Proline, compd., 1:1 Proline Co-crvstal , 1:1 Proline Co-crvstal …..…WO2011048112
CAS BASE 1291094-73-9, 416.46, C23 H28 O7
(1S)-1,5-Anhydro-1-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-D-glucitol bis[1-[(2S)-pyrrolidin-2-yl]ethanone]
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Sodium glucose transporter-2 inhibitor
SGLT 1/2 inhibitor
Novartis Ag innovator
Clinical trial……..https://clinicaltrials.gov/ct2/show/NCT01915849
https://clinicaltrials.gov/ct2/show/NCT02470403
- 10 Jun 2015 Novartis initiates enrolment in a phase II trial for Type 2 diabetes mellitus in USA (NCT02470403)
- 02 Apr 2014 Novartis terminates a phase II trial in Type-2 diabetes mellitus in USA, Poland, Argentina, Hungary, Puerto Rico and South Africa (NCT01824264)
- 01 Jan 2014 Novartis completes a phase II trial in Type 2 diabetes mellitus in USA (NCT01915849)


LIK-066 is in phase II clinical studies at Novartis for the treatment of type 2 diabetes.
In June 2014, the EMA’s PDCO adopted a positive opinion on a pediatric investigation plan (PIP) for LIK-066 for type 2 diabetes
Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition. Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
Type 1 diabetes, also known as Insulin Dependent Diabetes Mellitus (IDDM), is characterized by loss of the insulin-producing β-cells of the islets of Langerhans of the pancreas leading to a deficiency of insulin. Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) – is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin). Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from β-cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
Chronic elevation of blood glucose level also leads to damage of blood vessels. In diabetes, the resultant problems are grouped under “microvascular disease” (due to damage of small blood vessels) and “macro vascular disease” (due to damage of the arteries). Examples of microvascular disease include diabetic retinopathy, neuropathy and nephropathy, while examples of macrovascular disease include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
Diabetic retinopathy, characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US. Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy. Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide. A high glycemic diet (i.e., a diet that consists of meals that give high postprandial blood sugar) is known to be one of the causative factors contributing to the development of obesity.
Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level. Therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
Glucose is unable to diffuse across the cell membrane and requires transport proteins. The transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase. Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
SGLT belongs to the sodium/glucose co-transporter family SLCA5. Two different SGLT isoforms, SGLT1 and SGLT2, have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different. SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
As a consequence, glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments. Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule. In healthy human, more than 99% of plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1 % of the total filtered glucose being excreted in urine. It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule. A drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient’s needs for both blood glucose control, while preserving insulin secretion. In addition, SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
Indeed small molecule SGLT2 inhibitors have been discovered and the anti-diabetic therapeutic potential of such molecules has been reported in literature [T-1095 (Diabetes, 1999, 48, 1794-1800, Dapagliflozin (Diabetes, 2008, 57, 1723-1729)].
SYNTHESIS


PATENT
WO 2011048112https://www.google.com/patents/WO2011048112A1?cl=en
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
Patentscope, Espacenet
Example 61-62:

Example 61 : Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 mL), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Example 62: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 mL) and washed with brine (75 mL), brine containing 5 mL of 5% aqueous KHS04 (75 mL), and brine (20 mL) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.59)
H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J – 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m z 434.2 (M+18).
PICK UP IDEAS FROM HERE
Examples 57-58:

Step I: To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
Step II: To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2). The dichloromethane layers were combined and washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL X 2), and brine (200 mL), then dried over sodium sulfate and concentrated. The solid product was triturated with hexanes, and the triturated product was dried under vacuum to furnish (2-bromo-5-iodo-phenyl)-(2,3- dihydro-benzo[1 ,4]dioxin-6-yl)-methanone (30 g).
1H N R (400 MHz, DMSO-D6): δ 4.29-4.37 (m, 4H), 7.02 (d, J = 8.4 Hz, 1 H), 7.16 (d, J = 2.4 Hz, 1 H), 7.18-7.19 (m, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.77-7.81 (m, 1 H), 7.82 (d, J = 2.0 Hz, 1 H).
Step III: To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution (200 mL X 2), water (200 mL), and brine (200 mL), then dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish 6-(2-bromo-5-iodo-benzyl)-2,3- dihydro-benzo[1 ,4]dioxine (26.5 g). H NMR (400 MHz, DMSO-D6): δ 3.90 (s, 4H), 4.2 (s, 2H), 6.65 (dd, J = 8.4 Hz, J = 2.0 Hz, H), 6.68 (d, J = 2.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, H), 7.39 (d, J = 8.4 Hz, 1 H), 7.50 (dd, J = 8.4 Hz, J = 2.4 Hz 1 H), 7.67 (d, J = 2.8 Hz, 1 H).
Step IV: To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C. The reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C. After stirring for 1 h, 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq. NaHC03 solution (~75 mL) and extracted with ethyl acetate (250 mL X 3), dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish (3R,4S,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (28.4 g)
Example 57: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-1 ,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
Step V: To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C. After stirring for 4 h at 10°C, the reaction was quenched with saturated aqueous sodium bicarbonate (~ 100 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 X 150 mL). The organic layers were combined and dried over sodium sulfate, concentrated to furnish (3R,4R,5S,6R)-2- [4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol (28.4 g). Crude product was used for next reaction without purification. Example 58: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2!3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate Step V: To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature. After stirring for 2 h, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (500ml) and washed with 1 N HCI (200 mL X 2) followed by brine (200ml), then dried over sodium sulfate and
concentrated. The resulting crude compound was dissolved in ethanol (320 mL) at 65 °C and allowed to cool to room temperature while stirring. Light yellow solid formed was filtered and washed with cold ethanol (150 mL) followed by hexane (200 mL) to get acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin- 6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester powder (22.5 g, purity 98%).
COCRYSTAL
Example 75: 1:1 Proline Co-crvstal with f2S.3R.4R.5S.6R¾-2-r3-f2.3-Dihvdro- benzori.41dioxin-6-ylmethyl)-4-ethyl-phenvn-6-hvdroxymethyl-tetrahydro-pyran- 3.4.5-triol(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62) was completely amorphous initially but formed a crystalline complex with proline. This was confirmed by powder X-ray diffraction (PXRD) analysis. The stiochiometry of Example 62 and L- proline in the co-crystal prepared by method 1 was found to be 1 :1 by NMR
spectroscopy & HPLC. Characterization data for co-crystals of Example 62 and proline prepared by method 1 is shown in Table 3. Relative intensities of the most prominent powder x-ray diffraction peaks for co-crystals of Example 62 and proline are shown in Table 3A.
Table 3

Table 3A

3.70 15.78 18.36 25.18
9.68 10.68 18.88 36.33
11.07 21.21 20.42 69.29
14.26 14.81 21.18 27.94
14.80 22.97 22.50 12.25
15.40 4 98 23.78 33.08
16.12 8.45 24.56 6.92
16.59 18.78 25.79 21.69
17.31 100.0 27.46 8.90
17.60 20.35 31.97 7.65
17.98 47.20 32.46 5.98
1:1 Proline Co-crvstal
Example 77: 1:1 Proline Co-crvstal with (2S.3R.4R.5S.6Ri-2-f3-(2.3-Dihvdro- benzoh .41dioxin-6-ylmethvh-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triolMethod 2:
1 :1 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1500mg,3.6mmol), L- proline (415mg, 3.6mmol) and ethanol (23 ml_) were added to a 50 mL 3-neck round bottom flask equipped with nitrogen purging, magnetic stirring bar,
thermometer pocket & calcium chloride guard tube and the mixture was stirred at 25-30°C for 30 min., then heat to reflux. A clear solution was observed which was refluxed for 30 min., then slowly cool to 25-30°C causing percipitation. Di- isopropyl ether (DIPE, 23 mL) was added while maintaining the mixture at 25-30°C and stirring continuously for additional one to two hours at the same temperature. The precipitate was collected by filtration using vacuum (Nitrogen atmosphere), and the filter cake was washed with ethanol-DIPE mixture (1 :1 v/v, 10ml) followed by DIPE (23 mL). The product was vacuum dried at 65-70°C for 5-6 hrs.
1:1 Proline Co-crvstal (ΔΗ 53 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig. 1. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 2.
2:1 Proline Co-crvstal
Example 78: 2:1 Proline Co-crvstal with f2S.3R.4R.5S.6R>-2-r3-f2.3-Pihvdro-benzof1.41dioxin-6-ylmethvH-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triolMethod 3: 1 :2 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1 kg) was added to 15 L of ethanol with agitation while maintaining the mixture at 20-25 °C. The mixture was stirred for 10 min at 20-25 °C, then L-proline (537 gm) was added while maintaining the mixture at 20-25 °C. The mixture was stirred at this temperature for 30 min., then heated to reflux and refluxed for 30 min. The mixture was slowly cooled to 25-30°C then stired for 1 hr. DIPE (15 L) was added while maintaining the temperature at 25-30 °C and the mixture was stirred at this temperature for 1 hr. The precipitated product was collected by filtration and the product was washed with DIPE (5 L). The product was air dried at 65-70 °C to yield 1.22 kg
(79%) of a 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C (ΔΗ 85 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig.
3. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 4. Relative
intensities of the most prominent powder x-ray diffraction peaks for the 1 :2 co- crystals of Example 62 and proline are shown in Table 5.
Table 5

PATENT
WO 2012140597http://www.google.co.in/patents/WO2012140597A1?cl=en
. TABLE 2:

Intermediate 2: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-
 Intermediate 2
Intermediate 2
Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester
 To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1
,4]dioxin-6-ylmethyl)-
4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
(Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added
diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for
1 h at same temperature, reaction was quenched with the addition of 1 N
HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic
layers were washed with brine (10 ml), dried over sodium sulfate,
concentrated and purified by preparative HPLC to give 220 mg of
phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1
,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl
ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD):
δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J =
6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H),
3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H),
4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57
(dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m,
3H). MS (ES) m/z 553.3 (M+1 ).
To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1
,4]dioxin-6-ylmethyl)-
4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
(Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added
diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for
1 h at same temperature, reaction was quenched with the addition of 1 N
HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic
layers were washed with brine (10 ml), dried over sodium sulfate,
concentrated and purified by preparative HPLC to give 220 mg of
phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1
,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl
ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD):
δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J =
6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H),
3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H),
4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57
(dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m,
3H). MS (ES) m/z 553.3 (M+1 ).
Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

 To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1
,4]dioxin-6-
ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
(Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of
Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in
THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2
mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it
was stirred at room temperature for 20 min. Reaction mixture was cooled
to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5
ml). The reaction mixture was stirred at same temperature for 5 min and
then at room temperature for 2 h. Reaction mixture was cooled to 0°C and
quenched by the addition of 10% sodium bisulfite solution (5 ml). This
was extracted with ether (3 X 10 ml). Combined organic layer was washed
with brine (5 ml), dried over sodium sulfate and concentrated to give
700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-
[3-(2,3-dihydro-benzo[1
,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-
pyran-2-ylmethyl ester.
To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1
,4]dioxin-6-
ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
(Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of
Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in
THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2
mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it
was stirred at room temperature for 20 min. Reaction mixture was cooled
to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5
ml). The reaction mixture was stirred at same temperature for 5 min and
then at room temperature for 2 h. Reaction mixture was cooled to 0°C and
quenched by the addition of 10% sodium bisulfite solution (5 ml). This
was extracted with ether (3 X 10 ml). Combined organic layer was washed
with brine (5 ml), dried over sodium sulfate and concentrated to give
700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-
[3-(2,3-dihydro-benzo[1
,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-
pyran-2-ylmethyl ester.
To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.


Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester

Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.
PATENT
SEE INDIAN PATENT
IN 2009DE02173
Glycoside derivatives and uses thereof
REFERENCES
Pediatric investigation plan (PIP) decision: (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2:1) ( LIK066) (EMEA-001527-PIP01-13)European Medicines Agency (EMA) Web Site 2014, July 24
Safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) assessment of LIK066 in healthy subjects and in patients with type 2 diabetes mellitus (T2DM) (NCT01407003)
ClinicalTrials.gov Web Site 2011, August 07
WO2012140597
WO2011048112
IN 2009DE02173
| WO2001016147A1 | 24 Aug 2000 | 8 Mar 2001 | Kissei Pharmaceutical | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 | 2 Oct 2000 | 19 Apr 2001 | Bruce Ellsworth | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 | 15 Mar 2001 | 20 Sep 2001 | Hideki Fujikura | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 | 29 Mar 2001 | 11 Oct 2001 | Squibb Bristol Myers Co | O-aryl glucoside sglt2 inhibitors and method |
| WO2003020737A1 | 5 Sep 2002 | 13 Mar 2003 | Squibb Bristol Myers Co | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003043985A1 | 20 Nov 2002 | 30 May 2003 | Andrew Thomas Bach | Heterocyclic compounds and methods of use |
| WO2004018491A1 | 21 Aug 2003 | 4 Mar 2004 | Nobuhiko Fushimi | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004078163A2 | 26 Feb 2004 | 16 Sep 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004080990A1 | 12 Mar 2004 | 23 Sep 2004 | Kazuhiro Ikegai | C-glycoside derivatives and salts thereof |
| WO2004099230A1 | 30 Apr 2004 | 18 Nov 2004 | Eikyu Yoshiteru | Monosaccharide compounds |
| WO2004103995A1 | 19 May 2004 | 2 Dec 2004 | Gary Michael Ksander | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2005011592A2 | 29 Jul 2004 | 10 Feb 2005 | Janssen Pharmaceutica Nv | Substituted indazole-o-glucosides |
| WO2005021566A2 | 20 Aug 2004 | 10 Mar 2005 | Barsoumian Edward Leon | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085237A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085265A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006011502A1 | 27 Jul 2005 | 2 Feb 2006 | Chugai Pharmaceutical Co Ltd | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006054629A1 | 17 Nov 2005 | 26 May 2006 | Kissei Pharmaceutical | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2008016132A1 | 3 Aug 2007 | 7 Feb 2008 | Daiichi Sankyo Co Ltd | Benzyl phenyl glucopyranoside derivative |
| WO2011048112A1 * | 19 Oct 2010 | 28 Apr 2011 | Novartis Ag | Glycoside derivatives and uses thereof |
| US20030114390 * | 4 Oct 2002 | 19 Jun 2003 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| US20040018998 | 21 Sep 2001 | 29 Jan 2004 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US20060009400 | 28 Jun 2005 | 12 Jan 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948 | 15 Jul 2005 | 26 Jan 2006 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349 | 27 Jul 2005 | 2 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060035841 | 9 Aug 2005 | 16 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031 | 30 Sep 2005 | 6 Apr 2006 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060293252 | 14 Aug 2006 | 28 Dec 2006 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014 | 26 Jul 2007 | 31 Jan 2008 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015032272A1 * | 19 Aug 2014 | 12 Mar 2015 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9034921 | 1 Jun 2012 | 19 May 2015 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
INVENTORS OF LIK 066
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
| BEBERNITZ, Gregory, Raymond; (US). BOCK, Mark, G.; (US). REDDY, Dumbala Srinivas; (IN). HAJARE, Atul Kashinath; (IN). VYAVAHARE, Vinod; (IN). BHOSALE, Sandeep Bhausaheb; (IN). KURHADE, Suresh Eknath; (IN). SALUNKHE, Videsh; (IN). SHAIKH, Nadim, S.; (IN). BHUNIYA, Debnath; (IN). PALLE, P., Venkata; (IN). FENG, Lili; (US). LIANG, Jessica; (US) |
 Mark G Bock
Mark G BockBEBERNITZ, Gregory, Raymond….PIC NOT AVAILABLE
 Dr. Srinivasa Reddy
Dr. Srinivasa Reddy NADEEM SHAIKH
NADEEM SHAIKH Venkata Palle
Venkata PalleONLY FEW…………………….
//////see……..http://medcheminternational.blogspot.in/2015/11/lik-066-novartis-for-treatment-of-type.html
13
BEXAGLIFLOZIN



Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
Phase II
Developer:Theracos, Inc.
Indication:Type 2 diabetes
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)


Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.

Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative ("passive") glucose transporters (GLUTs) or sodium-dependent ("active") glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537...............PRODUCT PATENThttp://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)


Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694
Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
by crystallization of
((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
(1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and
water (1.3 kg) and the mixture was stirred until the solids dissolved.
The solution was filtered through filter membrane (Millipore, 0.45 μm)
into a clean glass reactor (50 L). The mixture was refluxed for 30 min
and water (7.2 kg) was added over 1.0 h while maintaining the
temperature between 50 and 65° C. The mixture was slowly cooled to ˜42°
C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.)
mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling
was continued to −5° C. over 12 h. The suspension was stirred for
another 5 h and was filtered. The solid was slurried with cold water and
filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under
reduced pressure for 24 h until the loss on drying was no more than 0.5%
to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in
25 mL THF) was added slowly dropwise over 30 min and the reaction
temperature was maintained below 10° C. After the addition was
completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol)
in THF (20 mL) was added dropwise over 30 min and kept the reaction
temperature below 10° C. After stirring for another 3 h at 20˜25° C.,
the reaction mixture was heated to reflux for additional 16 h and
monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled
to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl
acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL),
concentrated and the residue was purified by flash chromatography
(PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g
(70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A
100 mL of 4-necked flask equipped with thermometer and mechanical
stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1
mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane
(5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at
20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a
solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL)
was added dropwise over 30 min. After bubbling HBr for additional 2 h,
the mixture was refluxed for 3 days. The conversion was about 65%. The
mixture was quenched with ice water (50 mL), extracted with ethyl
acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL),
concentrated and the residue was purified by flash chromatography
(PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg
(52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4
Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95
(s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This
example describes the preparation of
2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling
of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl
4-methylbenzenesulfonate.

Under
nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg)
with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg,
0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg,
0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium
carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol).
After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that
starting material was consumed. The mixture was cooled to 20˜25° C.
The
reaction mixture was filtered over diatomite (28 kg) and the filter
cake was washed with acetone (2×31 kg). The combined filtrates were
transferred to a 500 L glass-lined reactor and concentrated. The residue
was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and
concentrated until the volume was about 100 L and was transferred to a
200 L glass-lined reactor and continued to concentrate to get about 22.5
kg of crude material.
The crude material was
dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min
with stirring (100 RPM) until it was a clear solution. The mixture was
cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The
suspension was stirred for another 5 h at 5 to 10° C. The mixture was
filtered at 0 to 5° C. and the filter cake was washed with pre-cooled
methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried
under at 15 to 20° C. for 15 h to give off-white to white solid. Yield:
18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3,
400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz,
2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H),
3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z):
429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example
9 Preparation of
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
bis(L-proline) complex
This example describes
preparation of
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
bis(L-proline) complex by co-crystallization of
((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
with L-proline in ethanol/water/n-heptane solvent mixture.
The
crude
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
(2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg)
and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While
keeping the temperature above 60° C., n-heptane (8.5 kg) was added over
40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at
this temperature for 10 h. The mixture was filtered and the solids were
washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L)
and the solids were dried under reduced pressure at 55 to 65° C. for 20 h
to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example
7 Preparation of
((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This
example describes preparation of
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
by removal of the anomeric OH or OMe.

A 30 L glass reactor equipped with a
thermometer was charged with crude
(2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
(1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was
magnetically stirred until all the solids dissolved under nitrogen
sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O
(1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane
(1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with
nitrogen sparging.
The cold
(2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
solution was added to the cold triethylsilane solution at such a rate
to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The
reaction mixture was stirred for another 2 to 3 h and then quenched by
addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg)
and the reaction mixture was stirred for about 15 min. The solvents
were removed under reduced pressure (2 h, temperature below 40° C.). The
residue was partitioned between ethyl acetate (6.9 kg) and water (3.9
kg). The layers were separated and the aqueous layer was extracted with
ethyl acetate (2×3.5 kg). The combined organic layers were washed with
brine (2×3.8 kg) and the solvents were removed under reduced pressure.
Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude
product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow
solid.
PATENT
WO 2011153953https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Preparation of 2-cyclopropoxyethanol (1)

Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2x15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
thanks for sharing with us.
ReplyDeleteCap Mockups