ALBACONAZOLE
Albaconazole (UR-9825) is a triazole antifungal. It has potential broad-spectrum activity.

ALBACONAZOLE

Albaconazole
Also known as: UNII-YDW24Y8IAB; UR-9825; 187949-02-6; UR 9825, W-0027
Molecular Formula: C20H16ClF2N5O2 Molecular Weight: 431.823146
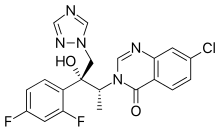
(1R,2R)-7-chloro-3-[2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]quinazolin-4(3H)-one
7-chloro-3-[(2R,3R)-3-(2,4-difluorophenyl)-3-hydroxy-4-(1,2,4-triazol-1-yl)butan-2-yl]quinazolin-4-one
Albaconazole (UR-9825) is a triazole antifungal. It has potential broad-spectrum activity.
Albaconazole is a broad-spectrum antifungal agent being evaluated in phase II clinical trials by Stiefel for the oral treatment of fungal infections, including toenail fungus, distal onychomycosis and subungual onychomycosis. Early clinical trials for the treatment of tinea pedis have been completed. In September 2005, Uriach, originator of albaconazole, granted Stiefel exclusive rights to develop and market albaconazole on a worldwide basis. In November 2006, Uriach’s R&D pipeline was transferred to Palau Pharma, a newly-created spin-out company. Under the terms of the agreement with Stiefel, Palau retains rights as comarketing partner in some European countries. In August 2013, Palau Pharma granted worldwide rights to Actavis. A triazole, albaconazole, has shown potent activity against a broad range of organisms, including pathogens resistant to other antifungals, such as fluconazole or itraconazole. It will be developed as an oral and topical formulation, and is expected to be available to the medical community for a variety of dermatological indications and fungal infections, including vulvovaginal candidiasis.
The condensation of the chiral oxazolidinone (I) with 2,4-difluorophenacyl bromide (II) by means of NaHMDS in THF/Et2 O gives the chiral oxirane (III), which is treated with LiOH and H2O2 to eliminate the chiral auxiliary, yielding the carboxylic acid (IV). The cleavage of the oxirane ring of (IV) with 1,2,4-triazole (V) and NaH in hot DMF affords the chiral hydroxyacid (VI), which is submitted to Curtius rearrangement by means of DPPA in hot pyridine to provide the chiral oxazolidinone (VII). The cleavage of the oxazolidinone ring of (VII) by means of refluxing aq. HCl gives the chiral aminoalcohol (VIII), which is condensed with 2-amino-4-chlorobenzoic acid (IX) by means of DCC and HOBt to yield the corresponding amide (X). Finally, this compound is cyclized to the target quinazolinone by reaction with triethyl orthoformate in hot dioxane/NMP.
The condensation of the chiral oxazolidinone (I) with 2,4-difluorophenacyl bromide (II) by means of NaHMDS in THF/Et2 O gives the chiral oxirane (III), which is treated with LiOH and H2O2 to eliminate the chiral auxiliary, yielding the carboxylic acid (IV). The cleavage of the oxirane ring of (IV) with 1,2,4-triazole (V) and NaH in hot DMF affords the chiral hydroxyacid (VI), which is submitted to Curtius rearrangement by means of DPPA in hot pyridine to provide the chiral oxazolidinone (VII). The cleavage of the oxazolidinone ring of (VII) by means of refluxing aq. HCl gives the chiral aminoalcohol (VIII), which is condensed with 2-amino-4-chlorobenzoic acid (IX) by means of DCC and HOBt to yield the corresponding amide (X). Finally, this compound is cyclized to the target quinazolinone by reaction with triethyl orthoformate in hot dioxane/NMP.
EP 0783501; ES 2107376; ES 2120885; JP 1998508317; US 5807854; WO 9705130
…………………………………………..

The condensation of (R)-lactic acid (I) with morpholine (II) gives the corresponding morpholide (III), which is protected at the hydroxyl position with dihydropyran (IV) to yield the tetrahydropyranyl ether (V). The Grignard reaction of (V) with 2,4-difluorophenylmagnesium bromide (VI) affords the chiral 1-propanone (VII), which by a Corey’s diastereoselective epoxidation with trimethylsulfoxonium iodide is converted into the oxirane (VIII). The opening of the oxirane ring of (VIII) by means of 1,2,4-triazole (IX) and NaH provides the tertiary alcohol (X), which is treated with pyridine p-toluenesulfonate to give the deprotected diol (XI) as a (2R,3R) and (2R,3S) 4:1 diastereomeric mixture, from which the desired (2R,3R)-isomer (XII) was isolated by crystallization. The reaction of (XII) with Ms-Cl and TEA, followed by cyclization with NaOMe, yields the oxirane (XIII), which is finally condensed with 7-chloroquinazolin-4(3H)-one (XIV) by means of K2CO3 in hot NMP.
ES 2159488; WO 0166519
…………………………………………….

Alternatively, intermediate (XIII) can be obtained as follows: Heating of ethyl (S)-lactate (XIV) with morpholine affords amide (XVI), which then reacts with 3,4-dihydro-2H-pyran (A) in the presence of p-TsOH to give protected derivative (XVII). Grignard reaction between (XVII), bromo derivative (XVIII) and Mg turnings in THF yields protected ketone (XIX), which is treated with pyridinium p-toluenesulfonate (PPTS) (THP group removal) and reprotected by means of Tf2O and DIEA to give triflate derivative (XX). Conversion of (XX) into intermediate (XIII) is achieved by reaction with triazolone (VII) and NaH in THF.
Chem Pharm Bull 1993,41(6),1035-42
……………………………………

Alternatively, derivative (XXIX) can be obtained in an analogous way as its enantiomer (XIX). Diastereoselective epoxidation of (XXIX) with trimethylsulfoxonium iodide and NaH in DMSO provides oxirane (XXX) (3). THP group removal by means of PPTS in EtOH, followed by reaction with 3,5-dinitrobenzoyl chloride (XXXI) and NaHCO3 in CH2Cl2, yields a diastereomeric mixture from which dinitrobenzoate derivative (2R,3R)-(XXXII) is obtained by recrystallization (1). Hydrolysis of (2R,3R)-(XXXII) in MeOH by treatment with aqueous NaOH gives compound (2R,3R)-(XXXIII), which is converted into ester (2R,3S)-(XXXIV) by Mitsunobu reaction with benzoic acid, Ph3P and DEAD in THF. Subsequent debenzoylation of (2R,3S)-(XXXIV) with NaOMe in MeOH affords oxiranyl ethanol derivative (2R,3R)-(XXXV), which is first converted into its triflate derivative by means of Tf2O and DIEA in CH2Cl2, and then into triazolone derivative (2S,3R)-(XXXVI) by reaction with intermediate (VII) and NaH in CH2Cl2/DMF. Finally, oxirane derivative (2S,3R)-(XXXVI) reacts with triazole (XXVI) and NaH in DMF to furnish the desired product.
…………………………………………………..

ER-30346 is synthesized by thiazole ring formation of (2R,3R)-3-(2,4-difluorophenyl)-3-hydroxy-2-methyl-4-(1H-1,2,4-triazol-1-yl)thiobutanamide (I) and 4-bromoacetylbenzonitrile (II) by means of reflux in methanol. The thioamide (I) is obtained with excellent yield from a chiral nitrile (III) by heating with diethyl dithiophosphate in aqueous medium.
…………………………………………….

The nitrile (III), a chiral key intermediate of this synthesis, can be obtained by two different synthetic routes as follows: Route-a: The key step of this route is ring opening reaction of the trisubstituted oxirane (VII) by cyanide anion leading to the nitrile (III). The chiral oxirane (VII) is synthesized from (R)-lactic acid derivatives as already reported. The reaction of (VII) with diethylaluminum cyanide in toluene or lithium cyanide in tetrahydrofuran gives the nitrile (III) with high yield without any epimerization reaction.
…………………………………………..

The nitrile (III), a chiral key intermediate of this synthesis, can be obtained by two different synthetic routes as follows: Route-b: The starting material of this route is methyl (S)-3-hydroxy-2-methylpropionate (VIII), which contains one additional carbon between the hydroxyl group and the 2-position carbon of (R)-lactate, the starting material of route-a. The hydroxyl group of (VIII) is protected by triphenylmethyl group. Then, 2,4-difluorophenyl moiety is introduced to give the ketone (X). Direct conversion of the ketone (X) to the oxirane (XIV) by dimethylsulfoxonium methylide, the same condition for compound (IV) in route-a, does not proceed. The oxirane (XIV) having desired stereochemistry is obtained via oxidation reaction. The ketone (X) is converted to the exomethylene (XI) by Wittig reaction. The stereoselective oxidation of (XI) is achieved by means of osmium tetroxide in the presence of 4-methylmorpholine N-oxide to give the diol (XII) in 58% yield after separation of its epimer by column chromatography. After methanesulfonylation of the primary alcohol of (XII), a triazole moiety is introduced and the triphenylmethyl group is deprotected. Then, the primary hydroxyl group of (XVI) is oxidized under Swern oxidation condition to give the aldehyde (XVII), which is converted to the chiral nitrile intermediate (III) by means of heating with hydroxylamine-O-sulfonic acid.
………………………………..
J. Med. Chem., 1998, 41 (11), pp 1869–1882
DOI: 10.1021/jm9707277
A series of azole antifungal agents featuring a quinazolinone nucleus have been subjected to studies of structure−activity relationships. In general, these compounds displayed higher in vitro activities against filamentous fungi and shorter half-lives than the structures described in our preceding paper. The most potent products in vitro carried a halogen (or an isostere) at the 7-position of the quinazolinone ring. Using a murine model of systemic candidosis, oral activity was found to be dependent on hydrophobicity, which, in turn, modulated the compound’s half-life. The 7-Cl derivative, (1R,2R)-7-chloro-3-[2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]quinazolin-4(3H)-one (20, UR-9825), was selected for further testing due to its high in vitro activity, low toxicity, good pharmacokinetic profile, and ease of obtention. Compound 20 is the (1R,2R) isomer of four possible stereoisomers. The other three isomers were also prepared and tested. The enantiomer (1S,2S) and the (1R,2S) epimer were inactive, whereas the (1S,2R) epimer retained some activity. In vitro 20 was superior to fluconazole, itraconazole, SCH-42427, and TAK-187 and roughly similar to voriconazole and ER-30346. In vivo, 20 was only moderately active in a mouse model of systemic candidosis when administration was limited to the first day. This was attributed to its short half-life in that species (t1/2 = 1 h po). Protection levels comparable to or higher than those of fluconazole, however, were observed in systemic candidosis models in rat and rabbit, where the half-life of the compound was found to be 6 and 9 h, respectively. Finally, 20 showed excellent protection levels in an immunocompromised rat model of disseminated aspergillosis. The compound showed low toxicity signs when administered to rats at 250 mg/kg qd or at 100 mg/kg bid during 28 days.
The condensation of the chiral oxazolidinone (I) with 2,4-difluorophenacyl bromide (II) by means of NaHMDS in THF/Et2 O gives the chiral oxirane (III), which is treated with LiOH and H2O2 to eliminate the chiral auxiliary, yielding the carboxylic acid (IV). The cleavage of the oxirane ring of (IV) with 1,2,4-triazole (V) and NaH in hot DMF affords the chiral hydroxyacid (VI), which is submitted to Curtius rearrangement by means of DPPA in hot pyridine to provide the chiral oxazolidinone (VII). The cleavage of the oxazolidinone ring of (VII) by means of refluxing aq. HCl gives the chiral aminoalcohol (VIII), which is condensed with 2-amino-4-chlorobenzoic acid (IX) by means of DCC and HOBt to yield the corresponding amide (X). Finally, this compound is cyclized to the target quinazolinone by reaction with triethyl orthoformate in hot dioxane/NMP.
J Med Chem 1998,41(11),1869
(1R,2R)-7-Chloro-3-[2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]quinazolin-4(3H)-one (20, UR-9825). Precipitated from EtOH/H2O (66% yield from amine 11): white amorphous solid;
mp 93−110 °C (wide range);
IR (KBr) ν 1675, 1601, 1554, 1498 cm-1;
1H NMR (300 MHz, CDCl3) 8.58 (s, 1H, N CH-N), 8.26 (d, J = 8.6, 1H, arom), 8.11 (d, J = 5.7, trace rotamer), 7.76 (s, 2H, triazol),
CH-N), 8.26 (d, J = 8.6, 1H, arom), 8.11 (d, J = 5.7, trace rotamer), 7.76 (s, 2H, triazol),
CH-N), 8.26 (d, J = 8.6, 1H, arom), 8.11 (d, J = 5.7, trace rotamer), 7.76 (s, 2H, triazol),
7.74 (d, J = 5.3, 1H, arom), 7.5 (m, 2H, arom), 7.10 (s, trace rotamer), 6.9−6.7 (m, 2H, arom),
5.91 (dq, Jd = 2, Jq = 7, 1H, MeCH), 5.54 (d, J = 2, 1H, OH),
5.15 (d, J = 14.2 1H, CH(H)), 4.9−4.7 (m, trace rotamer), 4.30 (d, trace rotamer), 3.99 (d, J = 14.2, 1H, CH(H)),
1.46 (d, J = 6.9, trace rotamer), 1.29 (d, J = 7, 3H, CHMe);
GC−MS 224 (Tr-CH2COHAr, C10H8F2N3O), 208 (group N-ethylheterocycle, C10H9ClN2O);
[α]D −8.0° (c 1, CHCl3).
Chiral HPLC (column, CicloBond SN 1; eluent, MeOH: Et3NHOAc in H2O at pH7 1:1; retention times: (S,S) (74) tR 12.6 min; (R,R) (20) tR 13.7 min). Area ratio: 0.01:99.99.
Anal. (C20H16ClF2N5O2) C, H, N.

 | |
| Systematic (IUPAC) name | |
|---|---|
7-Chloro-3-[(2R,3R)-3-(2,4-difluorophenyl)-3-hydroxy-4-(1,2,4-triazol-1-yl)butan-2-yl]quinazolin-4-one
| |
| Identifiers | |
| CAS Registry Number | 187949-02-6 |
| ATC code | None |
| PubChem | CID: 208952 |
| ChemSpider | 181045 |
| UNII | YDW24Y8IAB |
| KEGG | D09702 |
| ChEMBL | CHEMBL298817 |
| Chemical data | |
| Formula | C20H16ClF2N5O2 |
| Molecular mass | 431.823146 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।