
Belinostat (PXD101)
FAST TRACK FDA , ORPHAN STATUS
PXD101;PX105684;PXD
UNII:F4H96P17NZ
N-Hydroxy-3-(3-phen
N-HYDROXY-3-[3-[(PH
NSC726630

Approved by FDA......http://www.drugs.com/newdrugs/fda-approves-beleodaq-belinostat-peripheral-t-cell-lymphoma-4052.html?utm_source=ddc&utm_medium=email&utm_campaign=Today%27s+news+summary+-+July+3%2C+2014
July 3, 2014 -- The U.S. Food and Drug Administration today approved Beleodaq (belinostat) for the treatment of patients with peripheral T-cell lymphoma (PTCL), a rare and fast-growing type of non-Hodgkin lymphoma (NHL). The action was taken under the agency’s accelerated approval program.
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS...http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
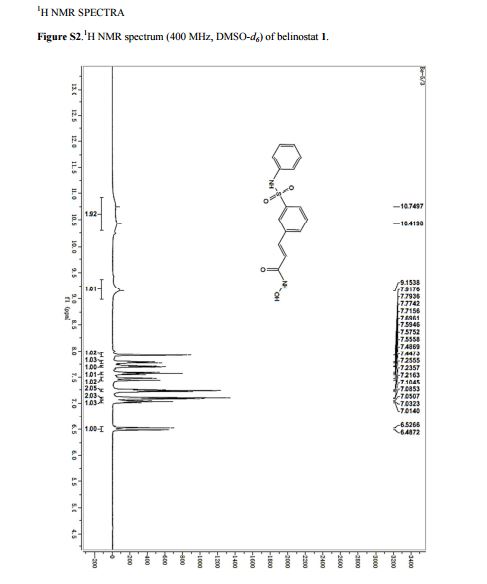
MP 172–174 °C, (lit.(@) 172 °C). 1H NMR (400 MHz, DMSO-d6) δ = 10.75–10.42 (m, 2H), 9.15 (s, 1H), 7.92 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H),7.47 (d, J = 15.8 Hz, 1H), 7.24 (m, 2H), 7.10–7.01 (m, 3H), 6.51 (d, J = 15.8 Hz, 1H). MS (ESI): m/z = 318.6 [M+H] +.
@ Finn, P. W.; Bandara, M.; Butcher, C.; Finn, A.; Hollinshead, R.; Khan, N.; Law, N.; Murthy, S.; Romero,R.; Watkins, C.; Andrianov, V.; Bokaldere, R. M.; Dikovska, K.; Gailite, V.; Loza, E.; Piskunova, I.;Starchenkov, I.; Vorona, M.; Kalvinsh, I. Helv. Chim. Acta 2005, 88, 1630, DOI: 10.1002/hlca.200590129
Beleodaq and Folotyn are marketed by Spectrum Pharmaceuticals, Inc., based in Henderson, Nevada. Istodax is marketed by Celgene Corporation based in Summit, New Jersey.
Belinostat was granted orphan drug status for the treatment of Peripheral T-cell lymphoma (PTCL) in the US in September 2009 and the EU in October 2012. In July 2015, an orphan drug designation has also been granted for malignant thymoma in the EU.
Belinostat received its first global approval in the US-FDA on 3 July 2014 for the intravenous (IV) treatment of relapsed or refractory PTCL in adults.
Belinostat was approved by the U.S. Food and Drug Administration (FDA) on July 3, 2014. It was originally developed by CuraGen Pharma,then developed by Spectrum Pharmaceuticals cooperating with Onxeo, then marketed as Beleodaq® by Spectrum.
Beleodaq is a pan-histone deacetylase (HDAC) inhibitor selectively causing the accumulation of acetylated histones and other proteinsin tumor cells. It is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
Beleodaq® is available as lyophilized powder for intravenous infusion, containing 500 mg of free Belinostat. The recommended dose is 1,000 mg/m2 once daily on days 1-5 of a 21-day cycle.


Beleodaq is a pan-histone deacetylase (HDAC) inhibitor selectively causing the accumulation of acetylated histones and other proteinsin tumor cells. It is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
Beleodaq® is available as lyophilized powder for intravenous infusion, containing 500 mg of free Belinostat. The recommended dose is 1,000 mg/m2 once daily on days 1-5 of a 21-day cycle.
Index:
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor

PTCL comprises a diverse group of rare diseases in which lymph nodes become cancerous. In 2014, the National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die. PTCL represents about 10 to 15 percent of NHLs in North America.Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.

Beleodaq works by stopping enzymes that contribute to T-cells, a type of immune cell, becoming cancerous. It is intended for patients whose disease returned after treatment (relapsed) or did not respond to previous treatment (refractory).
“This is the third drug that has been approved since 2009 for the treatment of peripheral T-cell lymphoma,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval expands the number of treatment options available to patients with serious and life-threatening diseases.”
The FDA granted accelerated approval to Folotyn (pralatrexate) in 2009 for use in patients with relapsed or refractory PTCL and Istodax (romidepsin) in 2011 for the treatment of PTCL in patients who received at least one prior therapy.
The safety and effectiveness of Beleodaq was evaluated in a clinical study involving 129 participants with relapsed or refractory PTCL. All participants were treated with Beleodaq until their disease progressed or side effects became unacceptable. Results showed 25.8 percent of participants had their cancer disappear (complete response) or shrink (partial response) after treatment.
The most common side effects seen in Beleodaq-treated participants were nausea, fatigue, fever (pyrexia), low red blood cells (anemia), and vomiting.
The FDA’s accelerated approval program allows for approval of a drug based on surrogate or intermediate endpoints reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs. Drugs receiving accelerated approval are subject to confirmatory trials verifying clinical benefit. Beleodaq also received orphan product designation by the FDA because it is intended to treat a rare disease or condition.
 BELINOSTAT
BELINOSTAT
Belinostat (trade name Beleodaq, previously known as PXD101) is a histone deacetylase inhibitor drug developed by TopoTargetfor the treatment of hematological malignancies and solid tumors.[2]
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin andpaclitaxel for relapsed ovarian cancer.[4] Final results in late 2009 of a phase II trial for T-cell lymphoma were encouraging.[5]Belinostat has been granted orphan drug and fast track designation by the FDA,[6] and was approved in the US for the use againstperipheral T-cell lymphoma on 3 July 2014.[3] It is not approved in Europe as of August 2014.[7]
The approved pharmaceutical formulation is given intravenously.[8]:180 Belinostat is primarily metabolized by UGT1A1; the initial dose should be reduced if the recipient is known to be homozygous for the UGT1A1*28 allele.[8]:179 and 181
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase


The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
 BELINOSTAT
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
CLIP
PXD101/Belinostat®(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.



PATENT
GENERAL SYNTHESIS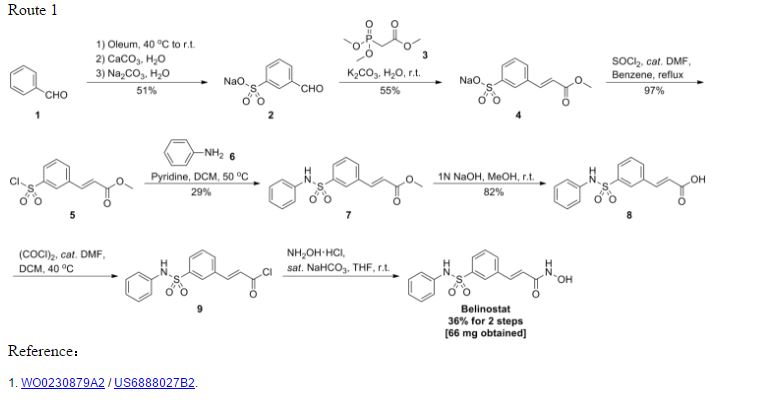
WO2002030879A2
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Scheme 3A






PATENT
SYNTHESISWO2002030879A2
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)


Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9x150 mm; mobile phase acetonitrile - 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
PATENT
https://www.google.com/patents/CN102786448A?cl=enExample: belinostat (compound of formula I) Preparation of

The compound of formula II (4. Og) added to the reactor, was added methanol 30ml, and stirred to dissolve, was added IM aqueous sodium hydroxide solution (38mL), stirred at room temperature overnight, the reaction was completed, ethyl acetate was added (IOmL) ^ K (20mL), stirred for 5 minutes, phase separation, the ethyl acetate phase was discarded, the aqueous phase was acidified with 10% hydrochloric acid to pH2, stirred at room temperature for 30 minutes, filtered, washed with water, and dried to give hydrolyzate 3. lg, yield rate of 81.6%.
The hydrolyzate (3. Og) added to the reactor, was added methylene chloride (53. 2g), dissolved with stirring, was added oxalyl chloride (2.8mL, 0.0032mol) at room temperature was added I drop DMF, reflux I hours, concentrated and the residue was dissolved in THF (30mL) alternate, the other to take a reaction flask was added hydroxylamine hydrochloride (3. 5g, 0.05mol), THF (50mL), saturated aqueous sodium bicarbonate (40mL), the mixture at room temperature under stirring for 10 minutes, then was added to spare, stirred at room temperature for I hour, the reaction was complete, at - at room temperature was added ethyl acetate (50mL), 2M hydrochloric acid (50mL), stirred for 5 minutes the phases were separated, the aqueous phase was discarded, the organic layer was washed with water, saturated brine, dried, filtered and concentrated to give crude product belinostat, recrystallized from ethyl acetate, 50 ° C and dried for 8 hours to give white crystals 2. 6g, yield 83.8%. . 1H-NMR (DMS0-d6, 400MHz) δ: 6 50 (1H, d, J = 16. OHz); 7 07 (d, J = 7. 8Hz, 2H); 7 16 (t.. , J = 7. 3Hz, 1H);. 7 25 (m, 2H);. 7 45 (t, J = 7. 8Hz, 1H);. 7 60 (d, J = 15. 9Hz, 1H); 7 . 62 (d, J = 7. 7Hz, 1H);. 7 75 (d, J = 7. 8Hz, 1H);. 7 88 (br s. '1H);. 9 17 (br s' 1H); 10. 35 (s, 1H);. 10 82ppm (br s, 1H). ·
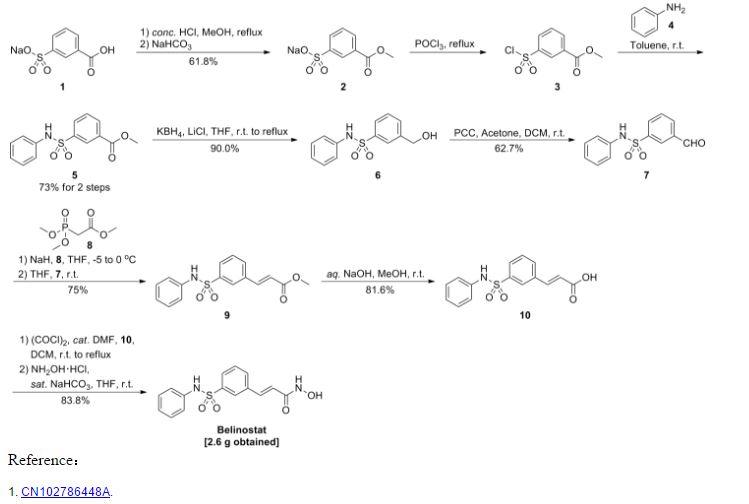
Step a): Preparation of Compound III

3-5 hours, filtered and the filtrate was added anhydrous sodium bicarbonate powder (200g), stirred for I hour, filtered, the filter residue was discarded, the filtrate was concentrated. The concentrate was added methanol (2000g), stirred at room temperature for 30 minutes, filtered and the filtrate was concentrated to dryness, 80 ° C and dried for 4 hours to give a white solid compound III147g, yield 61.8%.
Step b): Preparation of Compound IV

0-5 ° C, was slowly added to ice water, stirred for 2 hours and filtered to give a brown solid compound IV40 g, due to the instability of Compound IV, directly into the next reaction without drying.
Preparation of Compound V: [0040] Step c)

1H- bandit R (CDCl3, 400MHz) δ:.... 3 92 (s, 3H); 6 80 (. Br s, 1H); 7 06-7 09 (m, 2H); 7 11. . -7 15 (m, 1H);.. 7 22-7 26 (m, 2H);. 7 51 (t, J = 7. 8Hz, 1H);.. 7 90-7 93 (dt, J = . 1.2,7 8Hz, 1H); 8 18-8 21 (dt, J = I. 4, 7. 8Hz, 1H);... 8 48 (t, J = L 6Hz, 1H).
IR v ™ r: 3243,3198,3081,2953,1705,1438,1345,766,702,681cm-1.
Step d): Preparation of Compound VI

1H-NMR (DMS0-d6, 400ΜΗζ) δ:..... 4 53 (s, 2H); 5 39 (s, 1H); 6 99-7 03 (m, 1H); 7 08- 7. ll (m, 2H);.. 7 19-7 24 (m, 2H);.. 7 45-7 52 (m, 2H);.. 7 61-7 63 (dt, J = I. 8 , 7 4Hz, 1H);.. 7 79 (br s, 1H);. 10. 26 (s, 1H).
IRv =: 3453,3130,2964,1488,1151,1031, 757,688cm_10
Step e): Preparation of Compound VII

1H- bandit R (CDCl3,400MHz) δ:.... 7 08-7 15 (m, 4Η); 7 · 23-7 27 (m, 2H); 7 · 60-7 64 (t, J = 7 7Hz, 1Η);.. 8 00 (d, J = 7. 6Hz, 1Η);. 8 04 (d, J = 7. 6Hz, 1Η);. 8 30 (br s' 1Η).; 10. 00 (S, 1Η).
IR ν ™ Γ: 3213,3059,2964,2829,1687,1480,1348,1159,1082,758,679cm_10
Preparation of compounds of formula II: [0055] Step f)

1H-Nmr (Cdci3JOOmHz) δ:.... 3 81 (s, 3H); 6 40 (d, J = 16. 0Hz, 1H); 6 79 (. Br s, 1H); 7 08 ( d, J = 7. 8Hz, 2H);. 7 14 (t, J = 7. 3Hz, 1H);. 7 24 (m, 2H);. 7 46 (t, J = 7. 8Hz, 1H); 7. 61 (d, J = 16. ΟΗζ, ΙΗ);. 7 64 (d, J = 7. 6Hz, 1H);. 7 75 (d, J = 7. 8Hz, 1H);. 7 89 (br . s, 1H).
IR v ^ :: 3172,3081,2954,2849,1698,1475,1345,1157,773,714,677cm-1.
PATENT
SYNTHESISUS20100286279

CLIP
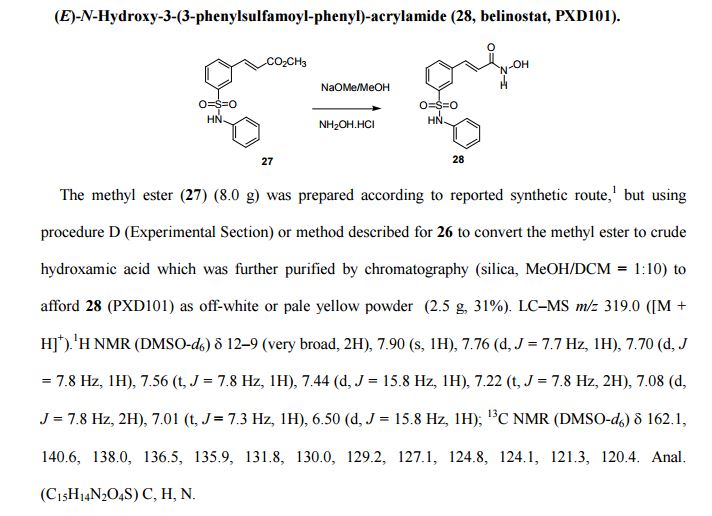
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 - 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1, 140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
(C15H14N2O4S) C, H, N
PATENT
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



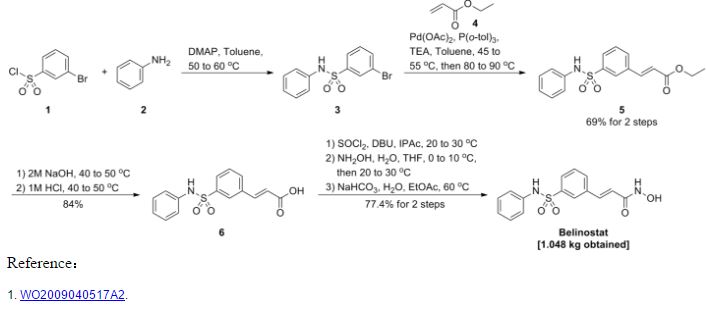
Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
PAPER
PATENT
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.

CLIP
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
CLIP
Copenhagen, December 10, 2013
Topotarget announces the submission of a New Drug Application (NDA) for belinostat for the treatment of relapsed or refractory (R/R) peripheral T-cell lymphoma (PTCL) to the US Food and Drug Administration (FDA). The NDA has been filed for Accelerated Approval with a request for Priority Review. Response from the FDA regarding acceptance to file is expected within 60 days from the FDA receipt date.
read all this here
PAPER
The Development of an Effective Synthetic Route of Belinostat
Key Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00170
Publication Date (Web): July 12, 2016
Copyright © 2016 American Chemical Society
*E-mail: guoliang222@gmail.com.

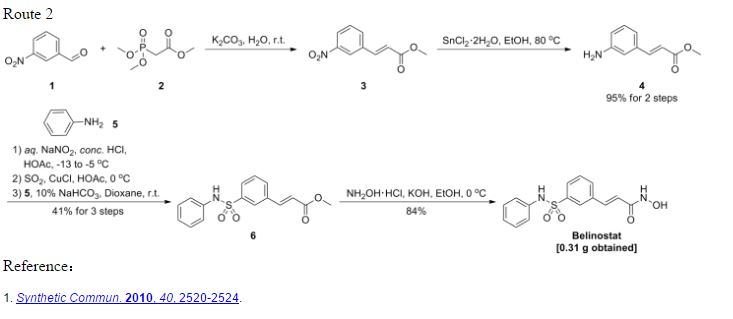
A practical synthetic route of belinostat is reported. Belinostat was obtained via a five-step process starting from benzaldehyde and including addition reaction with sodium bisulfite, sulfochlorination with chlorosulfonic acid, sulfonamidation with aniline, Knoevenagel condensation, and the final amidation with hydroxylamine. Key to the strategy is the preparation of 3-formylbenzenesulfonyl chloride using an economical and practical protocol. The main advantages of the route include inexpensive starting materials and acceptable overall yield. The scale-up experiment was carried out to provide 169 g of belinostat with 99.6% purity in 33% total yield.
(E)-N-Hydroxy-3-((phenylamino)sulfonyl)phenyl)acrylamide (Belinostat, 1)
1
mp 172–174 °C, (lit.(@) 172 °C). 1H NMR (400 MHz, DMSO-d6) δ = 10.75–10.42 (m, 2H), 9.15 (s, 1H), 7.92 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H),7.47 (d, J = 15.8 Hz, 1H), 7.24 (m, 2H), 7.10–7.01 (m, 3H), 6.51 (d, J = 15.8 Hz, 1H). MS (ESI): m/z = 318.6 [M+H] +.
@ Finn, P. W.; Bandara, M.; Butcher, C.; Finn, A.; Hollinshead, R.; Khan, N.; Law, N.; Murthy, S.; Romero,R.; Watkins, C.; Andrianov, V.; Bokaldere, R. M.; Dikovska, K.; Gailite, V.; Loza, E.; Piskunova, I.;Starchenkov, I.; Vorona, M.; Kalvinsh, I. Helv. Chim. Acta 2005, 88, 1630, DOI: 10.1002/hlca.200590129
Clip
Belinostat (Beleodaq ),
Belinostat is a drug which was developed by Spectrum Pharmaceuticals and is currently marketed by Onxeo as Beleodaq . The
drug, which received fast track designation by the United States Food and Drug Administration (US FDA) and was approved for
the treatment of hematological malignancies and solid tumors associated with peripheral T-cell lymphoma (PTCL) in 2014,58 is a histone deacetylase (HDAC) inhibitor and is the third such treatment to receive accelerated approval for PTCL, the others being
vorinostat (Zolinza ) and pralatrexate (Folotyn ).58 Although belinostat was not yet approved in Europe as of August 2014,58 the
compound exhibits a safety profile considered to be acceptable for HDAC inhibitors–less than 25% of patients reported adverse
effects and these most frequently were nausea, fatigue, pyrexia,anemia, and emesis.58 While several different synthetic approaches
have been reported for the preparation of belinostat and related HDAC inhibitors,59–62 the most likely process-scale approach has
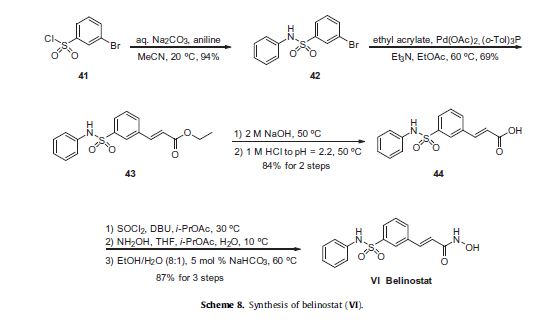
been described in a patent application filed by Reisch and co-workers at Topotarget UK, which exemplifies the synthesis described in
Scheme 8 on kilogram scale.63
drug, which received fast track designation by the United States Food and Drug Administration (US FDA) and was approved for
the treatment of hematological malignancies and solid tumors associated with peripheral T-cell lymphoma (PTCL) in 2014,58 is a histone deacetylase (HDAC) inhibitor and is the third such treatment to receive accelerated approval for PTCL, the others being
vorinostat (Zolinza ) and pralatrexate (Folotyn ).58 Although belinostat was not yet approved in Europe as of August 2014,58 the
compound exhibits a safety profile considered to be acceptable for HDAC inhibitors–less than 25% of patients reported adverse
effects and these most frequently were nausea, fatigue, pyrexia,anemia, and emesis.58 While several different synthetic approaches
have been reported for the preparation of belinostat and related HDAC inhibitors,59–62 the most likely process-scale approach has
been described in a patent application filed by Reisch and co-workers at Topotarget UK, which exemplifies the synthesis described in
Scheme 8 on kilogram scale.63
Commercially available 3-bromobenzenesulfonyl chloride (41) was reacted with aniline in the presence of aqueous sodium carbonate
to deliver sulfonamide 42 in 94% yield. Next, this aryl bromide was subjected to a Heck reaction involving ethyl acrylate to
give rise to cinnamate ester 43, which was immediately saponified under basic conditions and acidic workup to furnish the corresponding acid 44. This acid was activated as the corresponding acid chloride prior to subjection to hydroxylamine under basic conditions to form the hydroxamic acid, which was then recrystallized from an 8:1 ethanol/water mixture in the presence of a catalytic
amount of sodium bicarbonate to furnish crystalline belinostat (VI) in 87% overall yield from acid 44.61
to deliver sulfonamide 42 in 94% yield. Next, this aryl bromide was subjected to a Heck reaction involving ethyl acrylate to
give rise to cinnamate ester 43, which was immediately saponified under basic conditions and acidic workup to furnish the corresponding acid 44. This acid was activated as the corresponding acid chloride prior to subjection to hydroxylamine under basic conditions to form the hydroxamic acid, which was then recrystallized from an 8:1 ethanol/water mixture in the presence of a catalytic
amount of sodium bicarbonate to furnish crystalline belinostat (VI) in 87% overall yield from acid 44.61
Lee, H. Z.; Kwitkowski, V. E.; Del Valle, P. L.; Ricci, M. S.; Saber, H.;Habtemariam, B. A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X. H.;Brown, J.; Mehrotra, N.; Dorff, S.; Charlab, R.; Kane, R. C.; Kaminskas, E.;Justice, R.; Farrell, A. T.; Pazdur, R. Clin. Cancer Res. 2015, 21, 2666.
59. Qian, J.; Zhang, G.; Qin, H.; Zhu, Y.; Xiao, Y. CN Patent 102786448A, 2012.
60. Wang, H.; Yu, N.; Chen, D.; Lee, K. C.; Lye, P. L.; Chang, J. W.; Deng, W.; Ng, M.C.; Lu, T.; Khoo, M. L.; Poulsen, A.; ngthongpitag, K.; Wu, X.; Hu, C.; Goh, K.C.; Wang, X.; Fang, L.; Goh, K. L.; Khng, H. H.; Goh, S. K.; Yeo, P.; Liu, X.; Bonday, Z.; Wood, J. M.; Dymock, B. W.; Kantharaj, E.; Sun, E. T. J. Med. Chem.2011, 54, 4694.
61. Yang, L.; Xue, X.; Zhang, Y. Synth. Comm. 2010, 40, 2520.
59. Qian, J.; Zhang, G.; Qin, H.; Zhu, Y.; Xiao, Y. CN Patent 102786448A, 2012.
60. Wang, H.; Yu, N.; Chen, D.; Lee, K. C.; Lye, P. L.; Chang, J. W.; Deng, W.; Ng, M.C.; Lu, T.; Khoo, M. L.; Poulsen, A.; ngthongpitag, K.; Wu, X.; Hu, C.; Goh, K.C.; Wang, X.; Fang, L.; Goh, K. L.; Khng, H. H.; Goh, S. K.; Yeo, P.; Liu, X.; Bonday, Z.; Wood, J. M.; Dymock, B. W.; Kantharaj, E.; Sun, E. T. J. Med. Chem.2011, 54, 4694.
61. Yang, L.; Xue, X.; Zhang, Y. Synth. Comm. 2010, 40, 2520.
CLIP
Let's Research !!!!!

Helv Chim Acta 2005, 88(7), 1630-1657: It is first reported synthesis for Belinostat and many other derivatives. The procedure uses oleum, thionyl chloride (SOCl2) as well as oxalyl chloride (COCl)2, no wonder better procedures were derived from it. ABOVE
Synth Comm 2010, 40(17), 2520–2524: The synthesis avoids the use of the extremely corrosive oleum and thionyl chloride (SOCl2) and therefore is possibly better for scaled-up production. Second, synthetic steps do not involve tedious separations and give a better overall yield. BELOW Identifications:
Identifications:
Synth Comm 2010, 40(17), 2520–2524: The synthesis avoids the use of the extremely corrosive oleum and thionyl chloride (SOCl2) and therefore is possibly better for scaled-up production. Second, synthetic steps do not involve tedious separations and give a better overall yield. BELOW
Identifications:| 1H NMR (Estimated) for Belinostat |

Experimental: 1H NMR (300 MHz, DMSO-d6): δ 6.52 (d, J=15.9 Hz, 1H), 6.81–7.12 (m, 6H), 7.33 (d, J=15.9 Hz, 1H), 7.47–7.67 (m, 3 H), 7.87 (s, 1H), 9.00–11.20 (br, 3H).


SEE COMPILATION ON SIMILAR COMPOUNDS AT ..............http://drugsynthesisint.blogspot.in/p/nostat-series.html
HPLC
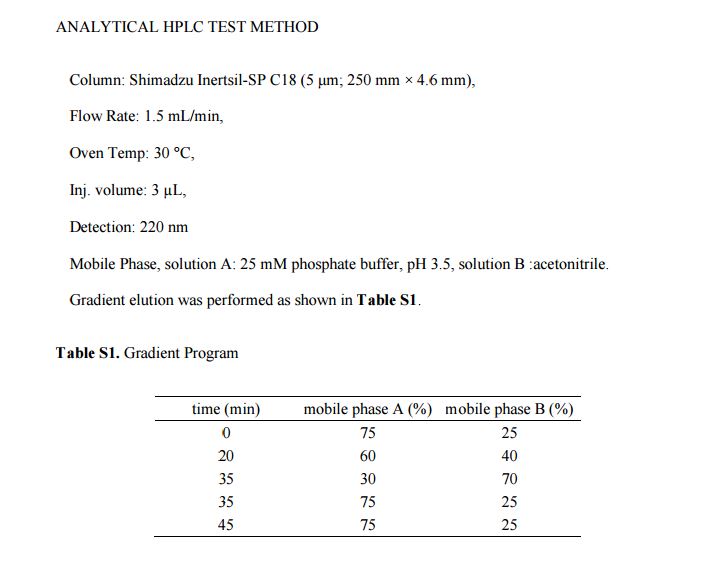
ANALYTICAL HPLC TEST METHOD
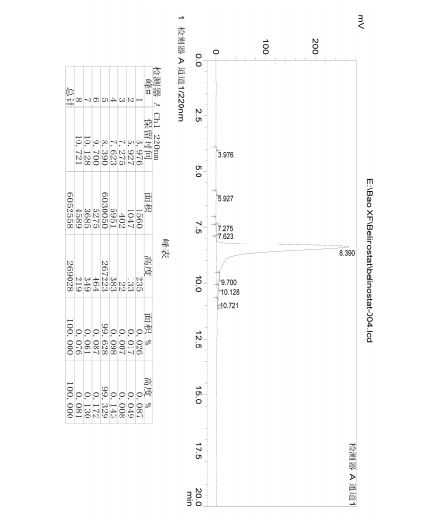
HPLC spectrum of Belinostat.
PATENT
Belinostat synthesis process related to the first report of the literature of W002 / 30879 A2, including preparation for Belinostat described as follows:


Example 3:
3- (3-sulfonate-yl) phenyl - acrylate preparation:
First, 3-bromophenyl sulfonate 37. Ig (257. 90g / mol, 0. 1439mol) was dissolved with stirring in 260mL toluene IL reactor was then added triethylamine 36. 5g (101. 19g / mol, 0. 3604mol), tri (o-methylphenyl) phosphine 0. 875g (304. 37g / mol, 0. 002874mol), palladium acetate 0. 324g (224. 51g, 0. 001441mol), the reaction mixture was heated to 45- 55 ° C with nitrogen pumping ventilation four, this time in the reaction system to generate the catalytically active 1 ^ (0). The temperature of the reaction system was raised to 80-90 ° C, within 2. 75h dropwise methacrylate 13. 6g (86. 04g / mol, 0. 1586mol), the reaction was continued after the cell by HPLC 3- bromophenyl sulfonyl chloride was completion of the reaction. The temperature of the reaction system was reduced to 45-55 ° C.
[0021] In at 45-55 ° C, the reaction mixture was concentrated under reduced pressure, ethyl acetate and n-heptane and recrystallized to give the product 29. 4g, 83% yield.
[0022] The spectral data:
1HNMR (DMS0-d6, HMDS0), δ (ppm): 3. 65 (3H, S, H-1); 6. 47 (1H, d, J = 16 0 Hz, H-2.); 7. 30 -8 00 (5H, m, H-3, H_4, H_5, H_6, H_7) m / e:. 264. 23

References
- "Beleodaq (belinostat) For Injection, For Intravenous Administration. Full Prescribing Information" (PDF). Spectrum Pharmaceuticals, Inc. Irvine, CA 92618. Retrieved 21 November2015.
- Plumb JA; Finn PW; Williams RJ; et al. (2003). "Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101". Molecular Cancer Therapeutics 2 (8): 721–728.PMID 12939461.
- "FDA approves Beleodaq to treat rare, aggressive form of non-Hodgkin lymphoma". FDA. 3 July 2014.
- "CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference". October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- "Spectrum adds to cancer pipeline with $350M deal.". February 2010.
- H. Spreitzer (4 August 2014). "Neue Wirkstoffe – Belinostat".Österreichische Apothekerzeitung (in German) (16/2014): 27.
- Lexicomp, (corporate author) (2016). Bragalone, DL, ed.Drug Information Handbook for Oncology (14th ed.). Wolters Kluwer. ISBN 9781591953517.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 - 1657, MP 172
- WO2009/40517 A2, ....
- WO2006/120456 A1, .....
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 - 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 - 4720, NMR IN SUP INFO
Biochem. J. 2008, 409, 581-589.
J. Transl. Med. 2007, 5, 1-12.
Mol. Cancer Ther. 2006, 5, 2086-2095.
Int. J. Cancer 2008, 122, 1400-1410.
. PLoS One 2013, 8, e54522.
Synthetic Commun. 2010, 40, 2520-2524.
| CN101868446A * | Sep 23, 2008 | Oct 20, 2010 | 托波塔吉特英国有限公司 | Methods of synthesis of certain hydroxamic acid compounds |
| CN102531972A * | Dec 31, 2010 | Jul 4, 2012 | 北京万全阳光医药科技有限公司 | Preparation method of intermediate of antitumor medicament Belinostat |
| EP2093292A2 * | Mar 26, 2001 | Aug 26, 2009 | Methylgene, Inc. | Inhibition of specific histone deacetylase isoforms |
| GB2378179A * | Title not available | |||
| WO2002030879A2 * | Sep 27, 2001 | Apr 18, 2002 | Prolifix Limited | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2008068170A1 * | Nov 27, 2007 | Jun 12, 2008 | William Paul Jackson | Hdac inhibitors |
| WO2009146871A1 * | Jun 1, 2009 | Dec 10, 2009 | William Paul Jackson | 5-lipoxygenase inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN104478769A * | Dec 22, 2014 | Apr 1, 2015 | 深圳万乐药业有限公司 | Belinostatsynthesis method suitable for industrial production |
| CN104478769B * | Dec 22, 2014 | Jan 6, 2016 | 深圳万乐药业有限公司 | 一种适合工业化生产的贝利司他合成方法 |
| CN104610100A * | Jan 9, 2015 | May 13, 2015 | 华东理工大学 | Nitrogen-chlorine type chlorination agent |
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| CN102786448A * | 9 avg 2012 | 21 nov 2012 | 深圳万乐药业有限公司 | Method of synthesizing belinostat |
| CN102786448B | 9 avg 2012 | 12 mar 2014 | 深圳万乐药业有限公司 | Method of synthesizing belinostat |
 | |
| Systematic (IUPAC) name | |
|---|---|
| (2E)-N-Hydroxy-3-[3-(phenylsulfamoyl)phenyl]prop-2-enamide | |
| Clinical data | |
| Trade names | Beleodaq |
| AHFS/Drugs.com | beleodaq |
| Pregnancy category |
|
| Routes of administration | Intravenous (IV) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Protein binding | 92.9–95.8%[1] |
| Metabolism | UGT1A1 |
| Excretion | Urine |
| Identifiers | |
| CAS Number | 866323-14-0 |
| ATC code | L01XX49 (WHO) |
| PubChem | CID 6918638 |
| ChemSpider | 5293831 |
| UNII | F4H96P17NZ |
| ChEBI | CHEBI:61076 |
| ChEMBL | CHEMBL408513 |
| Synonyms | PXD101 |
| Chemical data | |
| Formula | C15H14N2O4S |
| Molar mass | 318.348 g/mol |
////////////Belinostat, PXD101, novel HDAC inhibitor, Beleodaq, Folotyn, Spectrum Pharmaceuticals, Inc., Henderson, Nevada, Istodax, Celgene Corporation, Summit, New Jersey, CuraGen Pharma, FDA 2014
O=S(=O)(Nc1ccccc1)c2cc(\C=C\C(=O)NO)ccc2
SEE COMPILATION ON SIMILAR COMPOUNDS AT ..............http://drugsynthesisint.blogspot.in/p/nostat-series.html