

Baricitinib
NDA submitted jan 2016
NDA submitted jan 2016
2-[1-ethylsulfonyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile,
3-Azetidineacetonitrile, 1-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1Hpyrazol-1-yl]-
2-(3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile
2-(3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile
3-Azetidineacetonitrile, 1-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-
2-(3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile
For the treatment of rheumatoid arthritis and diabetic kidney disease
Incyte Corporation INNOVATOR
MF C16H17N7O2S
MW 371.4
SPONSOR Eli Lilly and Company
CODE LY3009104, INCB028050
CAS 1187594-09-7
WO 2009114512
MW 371.4
SPONSOR Eli Lilly and Company
CODE LY3009104, INCB028050
CAS 1187594-09-7
WO 2009114512
2-[3-(4-{7H-pyrrolo[2,3-d]pyrimidin-4-yl}-1H-pyrazol- 1-yl)-1-(ethylsulfonyl)azetidin-3-yl]acetonitrile (baricitinib) FREE FORM
m.p. 193–195 °C;
IR: 3203, 3113, 2998, 2847, 2363, 1584, 1328, 1137 cm–1.
Anal. calcd for C16H17N7 O2 S: C, 51.74; H, 4.61; N, 26.40; found: C, 51.91; H, 4.49; N, 26.57%. MS (m/z): 372 [M + H]+;
1 H NMR (300 MHz, DMSO-d6 ): δ 1.25 (t, J = 7.3 Hz, 3H), 3.23 (m, J = 7.3 Hz, 2H), 3.69 (s, 2H), 4.24 (d, J = 9.0 Hz, 2H), 4.61 (d, J = 9.0 Hz, 2H), 7.08 (s, 1H), 7.62 (s, 1H), 8.47 (s, 1H), 8.71 (s, 1H), 8.92 (s, 1H), 12.12 (s, 1H);
13C NMR (125 MHz, DMSO-d6 ): δ 7.4, 24.9, 39.3, 43.4, 58.5, 99.9, 113.0, 116.6, 126.9, 129.5, 139.9, 149.3, 150.9, 152.2.
REF Journal of Chemical Research, Volume 40, Number 4, April 2016,pp. 205-208(4)
![ChemSpider 2D Image | {1-(Ethylsulfonyl)-3-[4-(1H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-3-azetidinyl}acetonitrile phosphate (1:1) | C16H20N7O6PS](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_uTZaMmi6hpsNSZw_jIYhuhSu1dG0uqejCiC69G9MYGjUrlecQU2CBMMPO3ToWXLIQ69ARozqv2o3MCyQxQJKtWomapBo58_9l0bdJT-_awlWyWKYJZgX4Wb1YJz4hm97VIl6HpAwg4=s0-d)
Baricitinib phosphate
{1-(Ethylsulfonyl)-3-[4-(1H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-3-azetidinyl}acetonitrile phosphate (1:1)
1187595-84-1, C16H20N7O6PS, 469.41
- Originator Incyte Corporation
- Developer Eli Lilly; Incyte Corporation
- Class Acetonitriles; Antipsoriatics; Antirheumatics; Azetidines; Pyrazoles; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Janus kinase 1 inhibitors; Janus kinase-2 inhibitors
Highest Development Phases
- Preregistration Rheumatoid arthritis
- Phase II Atopic dermatitis; Diabetic nephropathies; Psoriasis; Systemic lupus erythematosus
Most Recent Events
- 01 Jul 2016 Eli Lilly completes a phase I trial in Healthy volunteers in China (PO) (NCT02758613)
- 09 Jun 2016 Efficacy and adverse events data from the RA-BEYOND phase III trial in Rheumatoid arthritis
The Janus kinase (JAK) is a family of four tyrosine receptor kinases that play a pivotal role in cytokine receptor signalling pathways via their interaction with signal transducers and
activators of transcription proteins. The four JAK family members are Janus kinase 1 (JAK1), Janus kinase 2 (JAK2), Janus kinase 3 (JAK3) and tyrosine kinase (TYK2), whose
lengths range from 120 to 140 kDa. It has been shown that JAK2 activation may be critical for tumour growth and progression,indicating its selection as a therapeutic target. Moreover, since JAK3 is required for immune cell development, targeting JAK3 could be a useful strategy for generating a novel class of immunosuppressant drugs. JAK1 and TYK2 have been implicated in disease and immune suppression.
activators of transcription proteins. The four JAK family members are Janus kinase 1 (JAK1), Janus kinase 2 (JAK2), Janus kinase 3 (JAK3) and tyrosine kinase (TYK2), whose
lengths range from 120 to 140 kDa. It has been shown that JAK2 activation may be critical for tumour growth and progression,indicating its selection as a therapeutic target. Moreover, since JAK3 is required for immune cell development, targeting JAK3 could be a useful strategy for generating a novel class of immunosuppressant drugs. JAK1 and TYK2 have been implicated in disease and immune suppression.
Over the past decade there have been extensive efforts to identify and design novel small-molecule JAK inhibitors with varied profiles of subtype selectivity to address unmet medical needs Ruxolitinib is a Janus kinase inhibitor with selectivity for subtypes JAK1 and JAK2. It was approved by the U.S. Food and Drug Administration (FDA) for the treatment
of intermediate or high-risk myelofibrosis in November 2011.
of intermediate or high-risk myelofibrosis in November 2011.
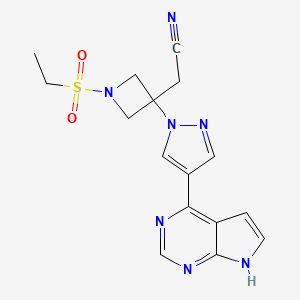
Selective inhibitors of JAK are viewed as having considerable potential as disease-modifying anti-inflammatory drugs for the treatment of rheumatoid arthritis. Tofacitinib, which was the first oral non-biological disease-modifying antirheumatic drug, was approved for the management of rheumatoid arthritis (RA) at the end of 2012. Baricitinib and filgotinib are beingmevaluated in phase III and phase II clinical trials respectively for the treatment of rheumatoid arthritis.
Baricitinib (also known as LY3009104 or INCB028050) is a novel and potent small molecule inhibitor of the Janus kinase family of enzymes with selectivity for JAK1 and JAK2. In in vitro studies baricitinib inhibited JAK1 and JAK2 in the low nanomolar range, while it demonstrated low inhibitory activity for JAK3 and moderate activity for TYK2.9–13 The data from two phase III studies showed that baricitinib can achieve impressive responses in RA patients who have not responded well to established therapies.
Therefore, improvement in the preparation of baricitinib is of practical significance.
1 L. Tan, K. Akahane, ET AL J. Med. Chem., 2015, 58, 6589.
2 J.J. Kulagowski, ET AL, J. Med. Chem., 2012, 55, 5901.
3 J.F. Kadow, ET AL , J. Med. Chem.,2012, 55, 2048.
4 Q. Su, ET AL J. Med. Chem., 2014, 57, 144.
5 J.D. Clark, ET AL J. Med. Chem., 2014, 57, 5023.
6 P. Norman, Expert. Opin. Investig. Drugs, 2014, 23, 1067.
7 L.J. Farmer, ET AL J. Med. Chem., 2015, 58,7195.
8 S.C. Meyer and R.L. Levine, Clin. Cancer. Res., 2014, 20, 2051.
4 Q. Su, ET AL J. Med. Chem., 2014, 57, 144.
5 J.D. Clark, ET AL J. Med. Chem., 2014, 57, 5023.
6 P. Norman, Expert. Opin. Investig. Drugs, 2014, 23, 1067.
7 L.J. Farmer, ET AL J. Med. Chem., 2015, 58,7195.
8 S.C. Meyer and R.L. Levine, Clin. Cancer. Res., 2014, 20, 2051.
Baricitinib (formerly INCB28050, LY3009104)[1] is an oral JAK1 and JAK2 inhibitor.
Baricitinib is in Phase III development by Eli Lilly and Incyte as a potential treatment for rheumatoid arthritis.[2] It is in Phase II development as a potential treatment for psoriasis and diabetic nephropathy. The related compound in JAK inhibitor is Tofacitinib, currently approved for the treatment of rheumatoid arthritis (RA) in the United States.
The companies announced 52-week data from a Phase IIb study of baricitinib at the European Congress of Rheumatology meeting in Madrid which showed that clinical improvements previously observed at week 24 were sustained for the full year in RA patients. Specifically, 49% of patients were ACR50 responders (ie a 50% improvement in their condition) after 52 weeks compared to 41% at week 24. For the full year, 21% reached ACR70 compared with 27% after 24 weeks.
To date, baricitinib, an orally administered selective JAK1 and JAK2 inhibitor, has demonstrated “an acceptable safety profile and side effects have generally been straightforward to manage”, said Oxford University’s Peter Taylor. He added that “these encouraging findings support further investigation of this new drug in RA”.
Baricitinib is already in Phase III for RA and in Phase II for psoriasis and diabetic nephropathy.
WO2009114512, also to Incyte, discloses azetidine and cyclobutane derivatives of the general structure shown below as JAK inhibitors.

Baricitinib (also known as LY3009104 or INCB28050) is in phase II clinical trials for the treatment of rheumatoid arthritis and diabetic kidney disease. Baricitinib is shown below.

About Baricitinib
Baricitinib is a once-daily, oral, selective JAK1 and JAK2 inhibitor. There are four known JAK enzymes: JAK1, JAK2, JAK3 and TYK2. JAK-dependent cytokines have been implicated in the pathogenesis of a number of inflammatory and autoimmune diseases, suggesting that JAK inhibitors may be useful for the treatment of a broad range of inflammatory conditions. Baricitinib demonstrates approximately 100-fold greater potency of inhibition against JAK1 and JAK2 than JAK 3 in kinase assays.
In December 2009, Lilly and Incyte announced an exclusive worldwide license and collaboration agreement for the development and commercialization of baricitinib and certain follow-on compounds for patients with inflammatory and autoimmune diseases. Baricitinib is currently in Phase 3 clinical development for rheumatoid arthritis and Phase 2 development for psoriasis and diabetic nephropathy.
About Rheumatoid Arthritis
Rheumatoid arthritis is an autoimmune diseasei characterized by inflammation and progressive destruction of joints.ii More than 23 million people worldwide suffer from RA.iii Approximately three times as many women as men have the disease. Patients and physicians indicate there remains an important opportunity to improve patient care. Current treatment of RA includes the use of non-steroidal anti-inflammatory drugs, oral disease-modifying anti-rheumatic drugs such as methotrexate, and injectable biological response modifiers that target selected mediators implicated in the pathogenesis of RA.iv
About Baricitinib Phase 3 Trials
Lilly and Incyte have conducted four pivotal Phase 3 clinical trials of baricitinib in patients with moderately-to-severely active rheumatoid arthritis to support regulatory submission in most countries. An additional Phase 3 study was initiated to support clinical development in China and remains ongoing. The clinical trial program includes a wide range of patients including those who are methotrexate naïve, inadequate responders to methotrexate, inadequate responders to conventional disease-modifying anti-rheumatic drugs, or inadequate responders to TNF inhibitors. Patients completing any of the five Phase 3 studies can enroll in a long-term extension study. For additional information on this clinical trial program, please visit http://www.clinicaltrials.gov.
About Incyte
Incyte Corporation is a Wilmington, Delaware-based biopharmaceutical company focused on the discovery, development and commercialization of proprietary therapeutics for oncology and inflammation. For additional information on Incyte, please visit the Company’s web site athttp://www.incyte.com.
About Eli Lilly and Company
Lilly is a global healthcare leader that unites caring with discovery to make life better for people around the world. We were founded more than a century ago by a man committed to creating high-quality medicines that meet real needs, and today we remain true to that mission in all our work. Across the globe, Lilly employees work to discover and bring life-changing medicines to those who need them, improve the understanding and management of disease, and give back to communities through philanthropy and volunteerism. To learn more about Lilly, please visit us at http://www.lilly.com and newsroom.lilly.com/social-channels.
SOURCE: Eli Lilly
Biological Activity
Baricitinib (formerly INCB28050, LY3009104) is a selective orally bioavailable JAK1/JAK2 inhibitor. Baricitinib preferentially inhibits JAK1 and JAK2, with 10-fold selectivity over Tyk2 and 100-fold over JAK3. INCB-28050 (baricitinib) inhibits intracellular signaling of multiple proinflammatory cytokines including IL-6 at concentrations <50 nM. Baricitinib also inhibits pSTAT3 stimulated by IL-23 with IC50 of 20 nM in isolated naive T-cells. Baricitinib (INCB028050) was also effective in multiple murine models of arthritis, with no evidence of suppression of humoral immunity or adverse hematologic effects. Baricitinib reduces levels of pSTAT3 in a dose- and time-dependent manner in the peripheral blood of rAIA animals. INCB28050 (Baricitinib) (10 mg/mL, p.o.) improves a composite score of joint damage by 47% in the murine CIA model.
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
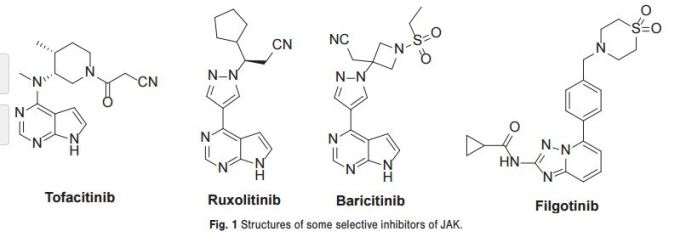
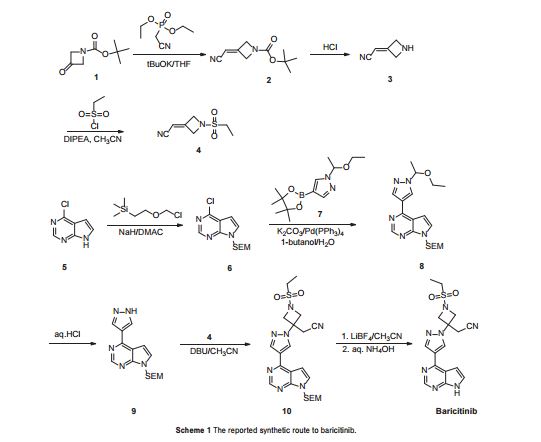
As shown in Scheme 1, Rodgers et al. have reported the first synthetic route to baricitinib.
ref J.D. Rodgers, S. Shepard, T.P. Maduskuie, H. Wang, N. Falahatpisheh, M. Rafalski, A.G. Arvanitis, L. Sorace, R.K. Jalluri, J.S. Fridman and K. Vaddi, 2007, US20070135461.
tert-Butyl 3-oxoazetidine- 1-carboxylate (1) was employed as the starting material. This was transformed to compound 2 by a Horner–Emmons reaction, followed by deprotection of the N-Boc group in acidic conditions. The intermediate 4 was obtained by the sulfonamidation reaction of compound 3 with ethanesulfonyl chloride. The other part of baricitinib was acquired by utilising 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (5) as the starting material. Compound 5 reacted with [2-(chloromethoxy)ethyl] trimethylsilane (SEM-Cl) to afford the intermediate 6, which was converted by reaction with 7 via the intermediate 8 to 4-(1H-pyrazol-4-yl)-7-{[2-(trimethylsilyl)ethoxy]methyl}-7Hpyrrolo[2,3-d]pyrimidine (9) via a Suzuki coupling reaction and a hydrolysis reaction. After the nucleophilic addition reaction and deprotection of the SEM group, baricitinib was obtained through eight steps. This synthetic route had drawbacks of high cost, low overall yield and the requirement of strict operating conditions.
PATENT
WO2009114512
EXAMPLES
Example 1. {l-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l- yl]azetidin-3-yl}acetonitrile trifluoroacetic acid salt

Step 1. tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate
0I \t
To a suspension of sodium hydride (60% dispersion in mineral oil, 0.257 g, 6.42 mmol) in tetrahydrofuran (32 mL) at 0 0C under a nitrogen atmosphere was added diethyl cyanomethylphosphonate (1.19 g, 6.72 mmol) (purchased from Aldrich). The reaction was then stirred for 45 minutes at room temperature. A solution of tert-butyl 3-oxoazetidine-l- carboxylate (1.00 g, 5.84 mmol) (purchased from Alfa Aesar) in tetrahydrofuran (8.8 mL) was introduced dropwise and the mixture was stirred for 16 hours. Brine and ethyl acetate were added and the layers separated. The aqueous layer was extracted with three portions of ethyl acetate. The combined extracts were dried over sodium sulfate, filtered and concentrated to afford product, used without further purification in Step 2 (1.12 g, 99%). 1H NMR (300 MHz, CDCl3): δ 5.38 (p, IH), 4.73-4.68 (m, 2H), 4.64-4.59 (m, 2H), 1.46 (s, 9H).
Step 2. tert-butyl 3-(cyanomethyl)’3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- djpyrim idin-4-yl) – 1 H-pyrazol-1 -yl]azetidine-l -carboxylate

To a solution of 4-(lH-pyrazol-4-yl)-7-[2-(trimethylsilyl)ethoxy]methyl-7H- pyrrolo[2,3-d]pyrimidine (4.61 g, 14.6 mmol) (prepared according to the method of WO 2007/070514 in Example 65, Step 2) and tert-butyl 3-(cyanomethylene)azetidine-l- carboxylate (2.84 g, 14.6 mmol) in acetonitrile (100 mL) was added 1,8- diazabicyclo[5.4.0]undec-7-ene (2.19 mL, 14.6 mmol). The reaction was stirred at room temperature for 16 hours. The acetonitrile was removed in vacuo and the residue was dissolved in ethyl acetate. This solution was sequentially washed with IN HCl and brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography, eluting with 80% ethyl acetate/hexanes to afford desired product (5.36 g, 72%).
1H NMR (300 MHz, CDCl3): δ 8.86 (s, IH), 8.44 (s, IH), 8.34 (s, IH), 7.42 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.54 (d, 2H), 4.29 (d, 2H), 3.59-3.51 (m, 2H), 3.33 (s, 2H), 1.47 (s, 9H), 0.96-0.89 (m, 2H), -0.06 (s, 9H); LCMS (M+H)+: 510.2.
Step 3. 3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH- pyrazol- 1 -yl] azetidin-3-ylacetonitrile

To a solution of tert-butyl 3-(cyanomethyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidine-l-carboxylate (5.36 g, 10.5 mmol) in 1,4-dioxane (100 mL) was added 4.00 M of hydrogen chloride in 1,4-dioxane (40 mL, 160 mmol) and the mixture was stirred at room temperature for 16 hours. The reaction was poured into saturated sodium bicarbonate solution sufficient to neutralize. The product was extracted with three portions of ethyl acetate. The combined extracts were washed with brine, dried over sodium sulfate, filtered and concentrated to afford product which was used without further purification (3.0 g, 69%). 1H NMR (400 MHz, CDCl3): δ 8.85 (s, IH), 8.42 (s, IH), 8.32 (s, IH), 7.41 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.30 (d, 2H), 3.88 (d, 2H), 3.58-3.51 (m, 2H), 3.42 (s, 2H), 0.96-0.89 (m, 2H), -0.06 (s, 9H); LCMS (M+H)+: 410.2. Step 4. l-(ethylsulfonyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- d]pyritnidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile

To a solution of 3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile (0.100 g, 0.244 mmol) in tetrahydrofuran (2 mL) containing N,N-diisopropylethylamine (0.085 mL, 0.49 mmol) was added ethanesulfonyl chloride (0.023 mL, 0.24 mmol). After stirring for 1.5 hours, the reaction mixture was poured into dilute HCl and extracted with three portions of ethyl acetate. The combined extracts were washed with brine, dried over sodium sulfate, decanted and concentrated to afford product, used without further purification in Step 5 (111 mg, 91%).
1H NMR (300 MHz, CDCl3): δ 8.86 (s, IH), 8.63 (s, IH), 8.35 (s, IH), 7.45 (d, IH), 6.83 (d, IH), 5.68 (s, 2H), 4.63 (d, 2H), 4.26 (d, 2H), 3.54 (t, 2H), 3.42 (s, 2H), 3.09 (q, 2H), 1.41 (t, 3H), 0.92 (t, 2H), -0.06 (s, 9H); LCMS (M+H)+: 502.1.
Step 5. l-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3- ylacetonitrile trifluoroacetate salt
To a solution of l-(ethylsulfonyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H- pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile (0.111 g, 0.22 mmol) in methylene chloride (3 mL) was added trifluoroacetic acid (2 mL) and the solution was stirred for 1.5 hours. The solvents were removed in vacuo and the residue was dissolved in methanol (3 mL) and ethylenediamine (0.1 mL) was added. After stirring for 3 hours, the volume was reduced in vacuo and the product was purified by preparative-HPLC/MS, (SunFire Cl 8 column, eluting with a gradient Of MeCNZH2O containing 0.1% TFA) to afford the product as the trifluoroacetic acid salt (50 mg, 47%). 1H NMR (400 MHz, d6-dmso): δ 12.55 (br d, IH), 9.03 (s, IH), 8.83 (s, IH), 8.56 (s, IH), 7.79-7.75 (m, IH), 7.24-7.19 (m, IH), 4.59 (d, 2H), 4.26 (d, 2H), 3.71 (s, 2H), 3.25 (q, 2H), 1.24 (t, 3H); LCMS (M+H)+: 372.1.
Alternatively, the deprotection and sulfonylation steps could be performed in the reverse order, as in Example 2.
Example 66. tert-Butyl 3-oxoazetidine-l-carboxylate (7).
A solution of tert-buty\ 3-hydroxyazetidine-l-carboxylate (24, 50 g, 289 mmol) in ethyl acetate (400 mL) was cooled to 0 0C. The resulting solution was then treated with solid TEMPO (0.5 g, 3.2 mmol, 0.011 equiv) and a solution of potassium bromide (KBr, 3.9 g, 33.2 mmol, 0.115 equiv) in water (60 mL) at 0 – 5 0C. While keeping the reaction temperature between 0 – 5 0C a solution of saturated aqueous sodium bicarbonate (NaHCO3, 450 mL) and an aqueous sodium hypochlorite solution (NaClO, 10 – 13 % available chlorine, 450 mL) were added. Once the solution of sodium hypochlorite was added, the color of the reaction mixture was changed immediately. When additional amount of sodium hypochlorite solution was added, the color of the reaction mixture was gradually faded. When TLC showed that all of the starting material was consumed, the color of the reaction mixture was no longer changed. The reaction mixture was then diluted with ethyl acetate (EtOAc, 500 mL) and two layers were separated. The organic layer was washed with water (500 rnL) and the saturated aqueous sodium chloride solution (500 mL) and dried over sodium sulfate (Na2SO4). The solvent was then removed under reduced pressure to give the crude product, tert-butyl 3-oxoazetidine-l-carboxylate (7, 48 g, 49.47 g theoretical, 97% yield), which was found to be sufficiently pure and was used directly in the subsequent reaction without further purification. For crude 7: 1H NMR (CDCl3, 300 MHz), δ 4.65 (s, 4H), 1.42 (s, 9H) ppm.
Example 67. tert-Buty\ 3-(cyanomethylene)azetidine-l-carboxylate (9). Diethyl cyanomethyl phosphonate (8, 745 g, 4.20 mol, 1.20 equiv) and anhydrous tetrahydrofuran (THF, 9 L) was added to a four-neck flask equipped with a thermowell, an addition funnel and the nitrogen protection tube at room temperature. The solution was cooled with an ice-methanol bath to -14 0C and a 1.0 M solution of potassium tert-butoxide (^-BuOK) in anhydrous tetrahydrofuran (THF, 3.85 L, 3.85 mol, 1.1 equiv) was added over 20 min while keeping the reaction temperature below -5 0C. The resulting reaction mixture was stirred for 3 h at -10 0C and a solution of l-terf-butoxycarbonyl-3-azetidinone (7, 600 g, 3.50 mol) in anhydrous tetrahydrofuran (THF, 2 L) was added over 2 h while keeping the internal temperature below -5 0C. The reaction mixture was stirred at -5 to -10 0C over 1 h and then slowly warmed up to room temperature and stirred at room temperature for overnight. The reaction mixture was then diluted with water (4.5 L) and saturated aqueous sodium chloride solution (NaCl, 4.5 L) and extracted with ethyl acetate (EtOAc, 2 x 9 L). The combined organic layers were washed with brine (6 L) and dried over anhydrous sodium sulfate (Na2SO4). The organic solvent was removed under reduced pressure and the residue was diluted with dichloromethane (CH2Cl2, 4 L) before being absorbed onto silica gel (Siθ2, 1.5 Kg). The crude product, which was absorbed on silica gel, was purified by flash column chromatography (SiO2, 3.5 Kg, 0 – 25% EtOAc/hexanes gradient elution) to afford tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate (9, 414.7 g, 679.8 g theoretical, 61% yield) as white solid. For 9: 1H NMR (CDCl3, 300MHz), δ 5.40 (m, IH), 4.70 (m, 2H), 4.61 (m, 2H), 1.46 (s, 9H) ppm; Ci0H14N2O2 (MW, 194.23), LCMS (EI) mle 217 (M+ + Na).
C8H13NO3 MoI Wt 171 19

2

11 step 3 10
C7H10N2O2S C5H7CIN2 MoI Wt 186 23 MoI Wt 130 58
Example 68. 2-(l-(Ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11).
A solution of tert-buty\ 3-(cyanomethylene)azetidine-l-carboxylate (9, 100Og, 5.2 mol) in acetonitrile (7 L) and a 3 N aqueous HCl solution (7 L) was stirred at room temperature for 18 h. When HPLC showed that all the starting material (9) was consumed, the reaction mixture was concentrated under reduced pressure to dryness. The residue, which contains the crude desired deprotection product (10), was then suspended in acetonitrile (12 L) and the resulting suspension was cooled to O – 5 0C. Diisopropyethylamine (DIEA, 3.14 L, 18.03 mol, 3.5 equiv) was then slowly added while keeping the internal temperature below 5 0C. The resulting homogeneous solution was allowed to cool down to O 0C and ethane sulfonyl chloride (EtSO2Cl, 730 mL, 7.73 mol, 1.5 equiv) was added over 1 h while keeping the internal temperature below 5 0C. The resulting reaction mixture was allowed to gradually warm to room temperature and stirred at room temperature for overnight. When HPLC showed that the reaction was complete, the reaction mixture was concentrated under reduced pressure to a volume of approximately 2 L. The bath temperature of the rotary evaporator is set to not exceed 45 0C. The concentrated residue was then diluted with dichloromethane (CH2CI2, 10 L) and the resulting dichloromethane solution was washed with aqueous sodium chloride solution (10 L). The aqueous phase was back extracted with dichloromethane (CH2CI2, 5 L). The combined organic layers were dried over anhydrous sodium sulfate
(Na2SO^ and the residue was absorbed onto silica gel (SiO2, 1 Kg) under reduced pressure. The bath temperature of the rotary evaporator was set to not exceed 45 0C. The material was then loaded onto a silica gel column (SiO2, 2.5 Kg) and eluted with 20 – 60 % ethyl acetate in heptane to afford 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 882 g, 968.4 g theoretical, 91 % yield) as off-white solids. For 11: 1H NMR (CDCl3, 300 MHz) δ 5.46 (m, IH), 4.77 (m, 2H), 4.70 (m, 2H), 3.05 (q, 2H), 1.39 (t, 3H) ppm; C7Hi0N2O2S (MW, 186.23), LCMS (EI) mle 187 (M+ + H).
Example 69. 2-(l-(Ethylsulfonyl)-3-(4-(7-((2-(trimethyIsiIyl)ethoxy)methyl)-7H- pyrrolo[2,3-</]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidin-3-yl)acetonitrile (12).
Method A. To a suspension of 4-(lH-pyrazol-4-yl)-7-((2-
(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-^pyrimidine (5, 440 g, 1.395 mol) and 2-(l- (ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 312.4 g, 1.68 mol, 1.2 equiv) in acetonitrile (4.4 L) was added DBU (249.8 mL, 1.67 mol, 1.2 equiv) drop wise to keep the reaction temperature between 15 – 25 0C. After adding DBU, the reaction mixture became homogeneous, but a precipitate appeared in 30 min. The reaction mixture was stirred for 3 h at room temperature. When ΗPLC showed that the reaction was deemed complete, the reaction mixture was quenched with water (11 L). The resulting mixture was stirred at room temperature for additional 30 min and then filtered. The solid cake was washed with water (4 L), MTBE (2 L) and dried in vacuum oven at 35 0C for 24 h to afford crude 2-(l –
(ethylsulfonyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-(i]pyrimidin-4-yl)- lH-pyrazol-l-yl)azetidin-3-yl)acetonitrile (12, 681 g, 699.8 g theoretical, 97.3 % yield) as white solids, which was found to be sufficiently pure for the subsequent reaction without further purification. For 12: 1HNMR (CDCl3, 300 MHz), δ 8.86 (s, IH), 8.45 (s, IH), 8.35 (s, IH), 7.43 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.65 (d, 2H), 4.27 (d, 2H), 3.55 (s, 2H), 3.4 (t, 2H), 3.07 (m, 2H), 1.42 (m, 3H), 0.92 (m, 2H), -0.05 (s, 9H) ppm; C22H3IN7O3SSi (MW, 501.68), LCMS (EI) mle 502 (M+ + H).

12
C15H21N5OSi C22H31N7O3SSi MoI Wt 315 45 MoI Wt 501 68

Phosphate salt
C16H17N7O2S C16H20N7O6PS
MoI Wt 371 42 MoI Wt 469 41
Example 72. tert-Butyl 3-(cyanomethyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrroIo[2,3-rf]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidine-l-carboxyIate (15).
To a suspension of tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate (9, 417.2 g, 2.15 mol, 1.05 equiv) and 4-(lH-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrrolo[2,3-<f]pyrimidine (5, 645 g, 2.04 mol) in acetonitrile (4.9 L) was added DBU (30.5 mL, 0.204 mol, 0.1 equiv) drop wise at room temperature. The resulting reaction mixture was then stirred at room temperature for 3 h. After about 1 h, a clear, brown solution was obtained. When LCMS showed that no starting material remained, silica gel (SiO2, 1 Kg) was added and the mixture was concentrated to dryness under reduced pressure. This material, which contains the crude desired product (15), was then loaded onto a pre-packed silica column (Siθ2, 2.5 Kg) and the column was eluted with 60 – 80% of ethyl acetate/heptane. The fractions containing the pure desired product (15) were combined and concentrated under reduced pressure to give the desired product as thick oil which was then stirred in heptane at room temperature until crystallization occurred. The solids were collected by filtration and washed with heptane to afford tert-buty\ 3-(cyanomethyl)-3-(4-(7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-ύ(]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidine- 1-carboxylate (15, 1014.9 g, 1039.7 g theoretical, 97.6% yield) as white solids. For 15: 1H NMR (DMSO-^6, 300 MHz) δ 8.93 (s, IH), 8.77 (s, IH), 8.47 (s, IH), 7.80 (d, IH, J= 3.8 Hz), 7.20 (d, IH, J = 3.7 Hz), 5.63 (s, 2H), 4.50 (d, 2H, J= 9.3 Hz), 4.21 (d, 2H, J= 9.3 Hz), 3.66 (s, 2H), 3.52 (t, 2H, J= 7.8 Hz), 1.40 (s, 9H), 0.82 (t, 2H, J= 8.1 Hz), -0.12 (s, 9H) ppm; C25H35N7O3Si (MW, 509.68), LCMS (EI) m/e 510 (M+ + H) and 532 (M+ + Na).

15
C15H21N5OSi C25H35N7O3Si MoI Wt 31545 MoI Wt 509 68

16 12
C20H27N7OSi C22H31N7O3SSi MoI Wt 409 56 MoI Wt 501 68

14 phosphate
C16H17N7O2S C16H20N7O6PS

MoI Wt 371 42 MoI Wt 46941
Example 77. (4-(l-(3-(Cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)- 7H-pyrrolo[2,3-</]pyrimidin-7-yl)methyl pivalate (20).
To a suspension of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-</|pyrimidin-7-yl]methyl pivalate (19, 10.0 g, 33.4 mmol) and 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 6.22 g, 33.4 mmol, 1.0 equiv) in N,N-dimethylformamide (DMF, 20 mL) was added DBU (254 mg, 1.67 mmol, 0.05 equiv) drop wise to keep the reaction temperature between 15 – 25 0C. After adding DBU, the reaction mixture became homogeneous within 90 min. The reaction mixture was stirred for 3 h at room temperature. When ΗPLC showed that the reaction was deemed complete, the reaction mixture was quenched with water (120 mL) and acetonitrile (80 mL). The resulting mixture was stirred at room temperature for an additional 30 min. The solids were collected by filtration, washed with a mixture of acetonitrile and water (2/3 by volume, 2 x 20 mL), and dried in vacuum oven at 40 – 45 0C for 24 h to afford crude (4-(l-(3-(cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)-7H- pyrrolo[2,3-</)pyrimidin-7-yl)methyl pivalate (20, 14.5 g, 16.2 g theoretical, 89.5 % yield) as white solids, which was found to be sufficiently pure (> 98.0% by ΗPLC) for the subsequent reaction without further purification. For 20: 1FTNMR (CDCl3, 300 MHz), δ 8.87 (s, IH), 8.43 (s, IH), 8.37 (s, IH), 7.51 (d, IH, J= 3.6 Hz), 6.76 (d, IH, J= 3.6 Hz), 6.26 (s, 2H),
4.64 (d, 2H, J = 9.6 Hz), 4.25 (d, 2H, J = 9.6 Hz), 3.41 (s, 2H), 3.09 (q, 2H, J= 7.6 Hz), 1.42 (t, 3H, J= 7.6 Hz), 1.17 (s, 9H) ppm; C22H27N7O4S (MW, 485.56), LCMS (EI) mle 486 (M+ + H).

C15H17N5O2 C22H27N7O4S MoI Wl 299 33 MoI Wt 48556

14 phosphate
C16H17N7O2S C16H20N7O6PS MoI Wt 371 42 MoI Wt 46941
PAPER
Authors: Xu, Jiaojiao; Cai, Jin; Chen, Junqing; Zong, Xi; Wu, Xuan; Ji, Min; Wang, Peng
Source: Journal of Chemical Research, Volume 40, Number 4, April 2016, pp. 205-208(4)
http://dx.doi.org/10.3184/174751916X14569294811333
A highly efficient method for the synthesis of baricitinib was developed. The starting material tert-butyl 3-oxoazetidine-1-carboxylate was converted to intermediate 2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile via the Horner–Emmons reaction, deprotection of the N-Boc-group
and a final sulfonamidation reaction. Then the nucleophilic addition
reaction was carried out smoothly to afford the borate intermediate in
the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene under reflux.
Finally, the desired compound baricitinib was obtained by the Suzuki
coupling reaction of 4-chloro-7-H-pyrrolo[2,3-d]pyrimidine with the above borate intermediate. All compounds were characterised by IR, MS, 1H NMR and 13C
NMR. The overall yield in this synthetic route was as high as 49%.
Moreover, this procedure is straightforward to carry out, has low cost
and is suitable for industrial production. http://dx.doi.org/10.3184/174751916X14569294811333
PATENT
Baricitinib is a Janus kinase (JAK) inhibitor. It is chemically designated as { 1 (ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile, having the structure as depicted in Formula I.

Formula I
U.S. Patent No. 8,158,616 discloses processes for the preparation of baricitinib of Formula I and [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.

Formula II
U.S. Patent No. 8, 158,616 involves a three-step process for the preparation of [4- (lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II as depicted in Scheme 1 below:
Scheme 1

Formula V

Formula VI

Formula II Formula VII
The process disclosed in U.S. Patent No. 8, 158,616 involves the use of sodium hydride as a base for reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III with chloromethyl pivalate of Formula IV, and the use of a protected pyrazole borolane derivative of Formula VI for the conversion of (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7- yl)methyl 2,2-dimethylpropanoate of Formula V into [4-(lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
The use of sodium hydride is not suitable on an industrial scale due to its inflammable and hazardous nature. The use of a protected pyrazole borolane derivative of Formula VI increases the cost of the manufacturing process, as an additional deprotection step is required for obtaining [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
Thus, there exists a need for the development of an economical and industrially advantageous process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II that avoids the use of sodium hydride and involves a lesser number of steps.
The present invention provides a convenient, economical, and industrially advantageous two-step process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II. The process of the present invention involves the use of an alkali or alkaline earth metal hydroxide, carbonate, or bicarbonate as a base for reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III with chloromethyl pivalate of Formula IV, and the use of an unprotected pyrazole borolane of Formula VIII for the conversion of (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate of Formula V into [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II. The process of the present invention provides [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II in high yield.
A first aspect of the present invention provides a process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II,

Formula II
comprising the steps of:
i) reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III

Formula III
with chloromethyl pivalate of Formula IV

Formula IV
in the presence of an alkali or alkaline earth metal hydroxide, carbonate, bicarbonate as a base to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7- yl)methyl 2,2-dimethylpropanoate of Formula V; and

ii) reacting the (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2- dimethylpropanoate of Formula V with 4-(4,4,5,5-tetramethyl-l,3,2 dioxaborolan-2-yl)-lH-pyrazole of Formula VIII

Formula VIII
to obtain the [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
A second aspect of the present invention provides a process for the preparation of baricitinib of Formula I,

Formula I
comprising the steps of:
i) reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III

Formula III
with chloromethyl pivalate of Formula IV

Formula IV
in the presence of an alkali or alkaline earth metal hydroxide, carbonate, or bicarbonate base to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate of Formula V;

Formula V
ii) reacting the (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2- dimethylpropanoate of Formula V with 4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-lH-pyrazole of Formula VIII

Formula VIII
to obtain [4-( lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II; and

Formula II
iii) reacting the [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II with [l-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile of Formula IX

Formula IX
to obtain baricitinib of Formula I.
EXAMPLES
Example 1 : Preparation of (4-chloro-7H-pyrrolor2.3-dlpyrimidin-7-yl)methyl 2.2-dimethylpropanoate (Formula V)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (25 g; Formula III), potassium carbonate (27 g), and chloromethyl pivalate (27 g; Formula IV) were added to a reaction vessel containing N,N-dimethylformamide (100 mL) at ambient temperature. The reaction mixture was stirred for 14 hours. The progress of the reaction was monitored by thin layer chromatography. Water (250 mL) was added to the reaction mixture, and then the mixture was stirred for 2 hours. The reaction mixture was filtered, then washed with water (50 mL), and then dried under reduced pressure at 40°C to 45°C for 12 hours to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate.
Yield: 98.85%
Example 2: Preparation of r4-(lH-pyrazol-4-yl)-7H-pyrrolor2.3-dlpyrimidin-7-yllmethyl pivalate (Formula II)
(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate (10 g; Formula V), water (50 mL), and potassium carbonate (15.5 g) were added into a reaction vessel at ambient temperature. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (8.7 g; Formula VIII), 1,4-dioxane (100 mL), and
tetrakis(triphenylphosphine)palladium(0) (0.08 g) were added to the reaction mixture. The reaction mixture was heated to a temperature of 80°C to 85°C, and then stirred at the same temperature for 14 hours. The progress of the reaction was monitored by thin layer chromatography. On completion, ethyl acetate (100 mL) was added to the reaction mixture. The contents were stirred for 1 hour, then filtered through a Hyflo®, and then washed with ethyl acetate (40 mL). The organic layer was separated, and then concentrated under reduced pressure to obtain [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate.
Yield: 82.27%
PATENT
WO-2014194195
PATENT
PATENT
The present invention provides processes for the preparation of baricitinib of Formula I and an intermediate of Formula V. The present invention also provides the of the intermediate of Formula V for the preparation of baricitinib.

Formula V
Background of the Invention
Baricitinib is a Janus kinase (JAK) inhibitor. It is chemically designated as (ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3 yl}acetonitrile, having the structure as depicted in Formula I.

Formula I
U.S. Patent No. 8,158,616 discloses a process for the preparation of baricitinib comprising the reaction of 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile of Formula II with 4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl methyl pivalate of Formula
III to provide an intermediate of Formula IV, followed by deprotection of the intermediate of Formula IV to obtain baricitinib of Formula I, as depicted in Scheme I below:
Scheme I

Formula IV
The process disclosed in U.S. Patent No. 8, 158,616 requires a deprotection step in the last stage of the synthesis, which adds to the cost of the overall synthesis.
Thus, there exists a need for an alternate, cost-effective, and industrially advantageous process for the preparation of baricitinib.
EXAMPLES
Example 1 : Preparation of 3-(cvanomethylene)azetidine hydrochloride (Formula VIP
Aqueous hydrochloric acid (6N, 10 mL) and montmorillonite K-10 (2 g) were added into a reaction vessel at ambient temperature. The contents were stirred for 1 hour, and then filtered under reduced pressure to obtain activated montmorillonite K-10. The activated montmorillonite K-10 was added into another reaction vessel containing tert-butyl 3-(cyanomethylidene)azetidine-l-carboxylate (2 g; Formula VI) and methanol (20
mL) at ambient temperature. The reaction mixture was refluxed for about 12 hours to about 15 hours. On completion, the reaction mixture was filtered under reduced pressure followed by recovery of methanol under reduced pressure at about 40°C to about 45°C to obtain 3-(cyanomethylene)azetidine hydrochloride.
Yield: 75%
Example 2: Preparation of 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (Formula ID
N,N-Diisopropylethylamine (4.5 mL) was added into a reaction vessel containing acetonitrile (50 mL) and 3-(cyanomethylene)azetidine hydrochloride (1.5 g; Formula VII) at about 0°C to about 10°C. The reaction mixture was stirred for about 10 minutes.
Ethanesulfonyl chloride (2.22 g) was added into the reaction mixture at about 0°C to about 5°C over about 5 minutes. The temperature of the reaction mixture was raised to about 20°C to about 25 °C, and then the reaction mixture was stirred for about 16 hours. On completion of the reaction, acetonitrile was recovered from the reaction mixture under reduced pressure at about 40°C to about 45°C to obtain an oily residue. Dichloromethane (50 mL) was added into the residue. The contents were washed with a saturated sodium chloride solution (30 mL), followed by complete recovery of dichloromethane under reduced pressure at about 40°C to obtain 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile .
Yield: 98.59%
Example 3: Preparation of { l-(ethylsulfonyl)-3-[4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-yl)-lH-pyrazol-l-yllazetidin-3-yl}acetonitrile (Formula V)
1,4-Dioxane (20 mL) was added into a reaction vessel containing a solution of potassium carbonate (4.5 g) in water (30 mL) at about 20°C to about 25 °C. 2-(l-(Ethylsulfonyl)azetidin-3-ylidene)acetonitrile (2 g; Formula II) and 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (2.30 g; Formula VIII) were added into the reaction mixture at about 20°C to about 25 °C. The reaction mixture was stirred at about 20°C to about 25 °C for about 16 hours to about 18 hours. On completion of the reaction, 1,4-dioxane was recovered from the reaction mixture under reduced pressure at about 45 °C to obtain a residue. Ethyl acetate (20 mL) was added into the residue, and the contents were stirred for about 5 minutes. The organic and aqueous layers were separated. The organic layer was concentrated under reduced pressure at about 45 °C to obtain { l-(ethylsulfonyl)- 3-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile.
Yield: 85.78%
Mass: 381.4 [M + H]+
Example 4: Preparation of baricitinib (Formula I)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (0.8 g; Formula IX) was added into a reaction vessel containing a solution of potassium carbonate (2.1 g) in water (30 mL) at about 20°C to about 25°C. A solution of { l-(ethylsulfonyl)-3-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile (2.0 g; Formula V) in 1,4-dioxane (30 mL) was added into the reaction mixture at about 20°C to about 25 °C, followed by the addition of tetrakis(triphenylphosphine)palladium(0) (0.1 g). The reaction mixture was stirred at about 80°C to about 85°C for about 5 hours. On completion of the reaction, 1,4-dioxane was recovered from the reaction mixture under reduced pressure at about 45°C to obtain a residue. Ethyl acetate (50 mL) was added into the residue, and then the contents were stirred for about 5 minutes. The organic and aqueous layers were separated. The organic layer was concentrated under reduced pressure at about 45°C to obtain baricitinib.
Yield: 99.0%
Patent
Figure 8 is a crystalline form II 1 the H NMR FIG.
PATENT
Figure 4: Infra-red (IR) spectrum of the crystalline form of baricitinib.
Example: Preparation of crystalline form of baricitinib
(4-( 1 -(3-(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3-yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate (8 g), methanol (40 mL), tetrahydrofuran (160 mL), and 1M sodium hydroxide (18.4 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched with water (80 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid. Half of the solvent was recovered at a temperature of 40°C to 50°C. The reaction mixture was stirred at 20°C to 25°C for 18 hours, and then cooled to 5°C to 10°C. The solids were filtered, washed with a mixture of acetonitrile (50 mL) and water (100 mL), and then dried at 40°C to 50°C under reduced pressure for 24 hours to obtain the crystalline form of baricitinib.
Yield: 70%
PATENT

Figure 1 : X-ray Powder Diffraction (XRPD) pattern of the crystalline form of baricitinib.
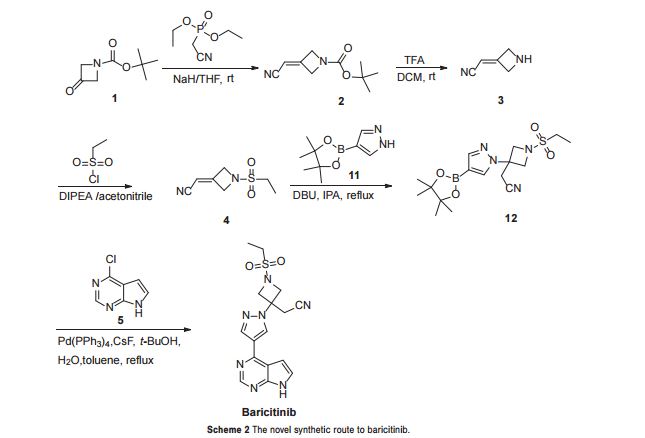
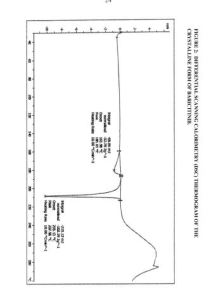 Figure 2: Differential Scanning Calorimetry (DSC) thermogram of the crystalline form of baricitinib.
Figure 2: Differential Scanning Calorimetry (DSC) thermogram of the crystalline form of baricitinib.
 Figure 3 : Thermogravimetric Analysis (TGA) of the crystalline form of baricitinib.
Figure 3 : Thermogravimetric Analysis (TGA) of the crystalline form of baricitinib.
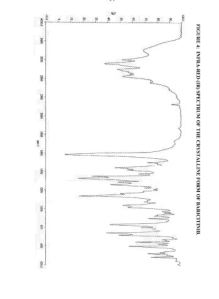 Figure 4: Infra-red (IR) spectrum of the crystalline form of baricitinib.
The crystalline form of baricitinib is further characterized by a DSC having endotherms at about 180.63°C and about 207.98°C.
The crystalline form of baricitinib has a water content of about 3%, as determined by TGA.
The crystalline form of baricitinib is also characterized by an XRPD pattern as depicted in Figure 1, a DSC thermogram as depicted in Figure 2, a TGA as depicted in Figure 3, and an IR spectrum as depicted in Figure 4.
The preparation of the crystalline form of baricitinib is carried out by reacting (4-(l-(3-(cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate with a base in the presence of one or more solvents at a temperature of about 15°C to 50°C, stirring the reaction mixture for about 30 minutes to about 10 hours, partially recovering the solvent(s) from the reaction mixture at a temperature of about 35°C to about 60°C under reduced pressure, stirring the contents at about 15°C to 35°C for about 5 hours to about 24 hours, filtering the solid, washing the solid with a mixture of acetonitrile and water, and drying.
The (4-( 1 -(3 -(cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate may be obtained by following the process disclosed in U.S. Patent No. 8, 158,616.
Figure 4: Infra-red (IR) spectrum of the crystalline form of baricitinib.
The crystalline form of baricitinib is further characterized by a DSC having endotherms at about 180.63°C and about 207.98°C.
The crystalline form of baricitinib has a water content of about 3%, as determined by TGA.
The crystalline form of baricitinib is also characterized by an XRPD pattern as depicted in Figure 1, a DSC thermogram as depicted in Figure 2, a TGA as depicted in Figure 3, and an IR spectrum as depicted in Figure 4.
The preparation of the crystalline form of baricitinib is carried out by reacting (4-(l-(3-(cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate with a base in the presence of one or more solvents at a temperature of about 15°C to 50°C, stirring the reaction mixture for about 30 minutes to about 10 hours, partially recovering the solvent(s) from the reaction mixture at a temperature of about 35°C to about 60°C under reduced pressure, stirring the contents at about 15°C to 35°C for about 5 hours to about 24 hours, filtering the solid, washing the solid with a mixture of acetonitrile and water, and drying.
The (4-( 1 -(3 -(cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate may be obtained by following the process disclosed in U.S. Patent No. 8, 158,616.
Example: Preparation of crystalline form of baricitinib
(4-( 1 -(3-(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3-yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate (8 g), methanol (40 mL), tetrahydrofuran (160 mL), and 1M sodium hydroxide (18.4 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched with water (80 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid. Half of the solvent was recovered at a temperature of 40°C to 50°C. The reaction mixture was stirred at 20°C to 25°C for 18 hours, and then cooled to 5°C to 10°C. The solids were filtered, washed with a mixture of acetonitrile (50 mL) and water (100 mL), and then dried at 40°C to 50°C under reduced pressure for 24 hours to obtain the crystalline form of baricitinib.
Yield: 70%



PATENT
EXAMPLES
Comparative Examples
Example 1 : Repetition of the process according to Example 78. Method B of U.S. Patent No. 8.158.616
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (1 g), methanol (5 mL), tetrahydrofuran (20 mL), and 1M sodium hydroxide (2.3 mL) were added into a reaction vessel at 20°C to 25 °C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched by adding water (20 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid, and the contents were stirred for 1.5 hours. No solid material was obtained. Example 2: Repetition of the process according to Example 78. Method C of U.S. Patent No. 8.158.616
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (2 g), lithium hydroxide monohydrate (0.51 g), acetonitrile (8 mL), and 2-propanol (2 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred at 45°C to 50°C for 6 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was cooled to 20°C to 25°C. The pH was adjusted to 6.0 to 7.0 by adding IN hydrochloric acid, and the contents were stirred overnight. No solid material was obtained.
Working Example:
Preparation of an amorphous form of baricitinib
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (1 g), methanol (5 mL), tetrahydrofuran (20 mL), and 1M sodium hydroxide (2.3 mL) were added into a reaction vessel at 20°C to 25 °C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched by adding water (20 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid, followed by completely recovering the solvent under reduced pressure at 40°C to 50°C. A sticky material was obtained. Water (10 mL) was added to the sticky material at 20°C to 25°C. The contents were stirred for 10 minutes. A solid material was precipitated out. The solid material was filtered, washed with water (20 mL), and then dried under reduced pressure at 40°C to 45°C for 24 hours to obtain the amorphous form of baricitinib.
Yield: 81%.
The amorphous form of baricitinib may be used in a pharmaceutical composition with one or more pharmaceutically acceptable carriers, diluents, or excipients, and optionally other therapeutic ingredients. The pharmaceutical composition may be used for the treatment of JAK-associated diseases.
PATENT
CN 105693731
PATENT
CN 105693731
Baricitinib crystal form A and preparation method
A process for prepn. of Baricitinib crystal form A is disclosed. The process comprises recrystn. of Baricitinib with DMF and water or alc., or ether to get the white powder Baricitinib crystal form A. The obtained crystal form A has high high-temp. stability, high-humidity stability, and light stability with XRPD spectrum at 2θ (±> 0.2) of 12.46, 13.921, 14.94, 15.359, 16.26, 16.639, 17.36, 19.08, 20.321, 21.961, 22.381, 24.118, 25.42, 27.441, 28.381, 29.321, 29.799, 32.675, 33.14, 33.563, 33.923, and 41.6. The Baricitinib crystal form A can be applied in the drugs for prevention and ............
Clip
A process for prepn. of Baricitinib crystal form A is disclosed. The process comprises recrystn. of Baricitinib with DMF and water or alc., or ether to get the white powder Baricitinib crystal form A. The obtained crystal form A has high high-temp. stability, high-humidity stability, and light stability with XRPD spectrum at 2θ (±> 0.2) of 12.46, 13.921, 14.94, 15.359, 16.26, 16.639, 17.36, 19.08, 20.321, 21.961, 22.381, 24.118, 25.42, 27.441, 28.381, 29.321, 29.799, 32.675, 33.14, 33.563, 33.923, and 41.6. The Baricitinib crystal form A can be applied in the drugs for prevention and ............
Clip
Crystalline forms of 1 ethylsulfonyl 3 4 7H pyrrolo 2 3 d pyrimidin 4 yl ...
priorart.ip.com/IPCOM/000244270
Nov 27, 2015 - Crystalline forms of baricitinib were found and are described ... on their appearance temperature As follows the polymorph observed at room ...
Mp. 213.8 °C (DSC).
IR (KBr): 3203, 3116, 2256, 1583, 1328, 1138 cm-1.
HNMR (DMSO-d6, 400 MHz): δ 12.17 (bs, 1H), 8.95 (s, 1H), 8.73 (s, 1H), 8.50 (s, 1H), 7.64
(d, J=3.2 Hz, 1H), 7.10 (d, J=3.4 Hz, 1H), 4.62 (d, J=9.0 Hz, 2H), 4.26 (d, J=9.1 Hz,
2H), 3.72 (s, 2H), 3.26 (q, J=7.3 Hz, 2H), 1.26 (t, J=7.3 Hz, 3H) ppm.
CNMR (DMSO-d6, 100 MHz): δ 152.39, 151.10, 149.55, 140.10, 129.80, 127.13, 122.42,
116.86, 113.25, 100.14, 58.74, 56.26, 43.50, 27.03, 7.63 ppm.
HSQC (optimized for JC-H = 145 Hz): 8.95-129.80, 8.73-151.10, 8.50-140.10, 7.64-127.13,
7.10-100.14, 4.62-58.74, 4.26-58.74, 3.72-27.03, 3.26-43.50, 1.26-7.63.
HMBC (optimized for JC-H = 8 Hz): 12.17-(127.13, 113.25, 100.14), 8.95-(140.10, 122.42,
56.26), 8.73-(152.39, 149.55, 113.25), 8.50-(129.80, 122.42), 7.64-(152.39, 113.25,
100.14), 7.10-(152.39, 127.13, 113.25), (4.62, 4.26)-(58.74, 56.26, 27.03), 3.72-
(116.86, 58.74, 56.26), 3.26-7.63, 1.26-43.50.
Calcd. C16H17N7O2S (M 371.42):
C 51.74%; H 4.61%; N 26.40%; S 8.63%.
Found C 51.62%; H 4.59%; N 26.28%; S 8.78%.
priorart.ip.com/IPCOM/000244270
Nov 27, 2015 - Crystalline forms of baricitinib were found and are described ... on their appearance temperature As follows the polymorph observed at room ...
Mp. 213.8 °C (DSC).
IR (KBr): 3203, 3116, 2256, 1583, 1328, 1138 cm-1.
IR (KBr): 3203, 3116, 2256, 1583, 1328, 1138 cm-1.
HNMR (DMSO-d6, 400 MHz): δ 12.17 (bs, 1H), 8.95 (s, 1H), 8.73 (s, 1H), 8.50 (s, 1H), 7.64
(d, J=3.2 Hz, 1H), 7.10 (d, J=3.4 Hz, 1H), 4.62 (d, J=9.0 Hz, 2H), 4.26 (d, J=9.1 Hz,
2H), 3.72 (s, 2H), 3.26 (q, J=7.3 Hz, 2H), 1.26 (t, J=7.3 Hz, 3H) ppm.
CNMR (DMSO-d6, 100 MHz): δ 152.39, 151.10, 149.55, 140.10, 129.80, 127.13, 122.42,
116.86, 113.25, 100.14, 58.74, 56.26, 43.50, 27.03, 7.63 ppm.
HSQC (optimized for JC-H = 145 Hz): 8.95-129.80, 8.73-151.10, 8.50-140.10, 7.64-127.13,
7.10-100.14, 4.62-58.74, 4.26-58.74, 3.72-27.03, 3.26-43.50, 1.26-7.63.
7.10-100.14, 4.62-58.74, 4.26-58.74, 3.72-27.03, 3.26-43.50, 1.26-7.63.
HMBC (optimized for JC-H = 8 Hz): 12.17-(127.13, 113.25, 100.14), 8.95-(140.10, 122.42,
56.26), 8.73-(152.39, 149.55, 113.25), 8.50-(129.80, 122.42), 7.64-(152.39, 113.25,
100.14), 7.10-(152.39, 127.13, 113.25), (4.62, 4.26)-(58.74, 56.26, 27.03), 3.72-
(116.86, 58.74, 56.26), 3.26-7.63, 1.26-43.50.
Calcd. C16H17N7O2S (M 371.42):
C 51.74%; H 4.61%; N 26.40%; S 8.63%.
Found C 51.62%; H 4.59%; N 26.28%; S 8.78%.
References
- “Baricitinib” (pdf). Statement on a nonproprietary name adopted by the USAN council. American Medical Association.
- “Lilly, Incyte Treatment Shows Positive Results”.http://www.insideindianabusiness.com. 9 Dec 2014. Retrieved 2 Mar 2015.
| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| AZETIDINE AND CYCLOBUTANE DERIVATIVES AS JAK INHIBITORS [US8158616] | 2009-09-17 | 2012-04-17 |
| AZETIDINE AND CYCLOBUTANE DERIVATIVES AS JAK INHIBITORS [US2013225556] | 2013-03-29 | 2013-08-29 |
| JANUS KINASE INHIBITORS FOR TREATMENT OF DRY EYE AND OTHER EYE RELATED DISEASES [US2010113416] | 2010-05-06 | |
| METHOD OF TREATING MUSCULAR DEGRADATION [US2013310340] | 2013-05-15 | 2013-11-21 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| Azetidine and cyclobutane derivatives as JAK inhibitors [US8420629] | 2011-12-09 | 2013-04-16 |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| CITING PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|---|---|---|---|
| WO2010039939A1* | Oct 1, 2009 | Apr 8, 2010 | Incyte Corporation | Janus kinase inhibitors for treatment of dry eye and other eye related diseases |
| WO2011028685A1 | Aug 31, 2010 | Mar 10, 2011 | Incyte Corporation | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| WO2011103423A1* | Feb 18, 2011 | Aug 25, 2011 | Incyte Corporation | Cyclobutane and methylcyclobutane derivatives as janus kinase inhibitors |
| WO2012068450A1* | Nov 18, 2011 | May 24, 2012 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as jak inhibitors |
| WO2012177606A1 | Jun 19, 2012 | Dec 27, 2012 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as jak inhibitors |
| WO2013026025A1 | Aug 17, 2012 | Feb 21, 2013 | Incyte Corporation | Cyclohexyl azetidine derivatives as jak inhibitors |
| WO2013173506A2 | May 15, 2013 | Nov 21, 2013 | Rigel Pharmaceuticals, Inc. | Method of treating muscular degradation |
| WO2014028756A1* | Aug 15, 2013 | Feb 20, 2014 | Concert Pharmaceuticals, Inc. | Deuterated baricitinib |
| WO2014138168A1* | Mar 5, 2014 | Sep 12, 2014 | Incyte Corporation | Processes and intermediates for making a jak inhibitor |
| WO2015095492A1 | Dec 18, 2014 | Jun 25, 2015 | Incyte Corporation | Tricyclic heterocycles as bet protein inhibitors |
| WO2015123424A1 | Feb 12, 2015 | Aug 20, 2015 | Incyte Corporation | Cyclopropylamines as lsd1 inhibitors |
| WO2015131031A1 | Feb 27, 2015 | Sep 3, 2015 | Incyte Corporation | Jak1 inhibitors for the treatment of myelodysplastic syndromes |
| CN102844317B * | Feb 18, 2011 | Jun 3, 2015 | 因西特公司 | Cyclobutane and methylcyclobutane derivatives as janus kinase inhibitors |
| CN103415515B * | Nov 18, 2011 | Aug 26, 2015 | 因塞特公司 | 作为jak抑制剂的环丁基取代的吡咯并吡啶和吡咯并嘧啶衍生物 |
| CN103797010A * | Jun 19, 2012 | May 14, 2014 | 因塞特公司 | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| CN104024256A * | Sep 6, 2012 | Sep 3, 2014 | 因塞特公司 | Processes and intermediates for making a JAK inhibitor |
| EP2647629A1 * | Dec 1, 2011 | Oct 9, 2013 | Nissan Chemical Industries, Ltd. | Pyrazole compound having therapeutic effect on multiple myeloma |
| EP2647629A4 * | Dec 1, 2011 | Apr 23, 2014 | Nissan Chemical Ind Ltd | Pyrazole compound having therapeutic effect on multiple myeloma |
| US8415362 | Jun 12, 2008 | Apr 9, 2013 | Incyte Corporation | Pyrazolyl substituted pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8722693 | Dec 5, 2013 | May 13, 2014 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8822481 | Apr 18, 2014 | Sep 2, 2014 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8829013 | Apr 18, 2014 | Sep 9, 2014 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8933085 | Nov 18, 2011 | Jan 13, 2015 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US8933086 | Sep 20, 2013 | Jan 13, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B]pyridines and pyrrolo[2,3-B]pyrimidines as Janus kinase inhibitors |
| US8946245 | Mar 30, 2011 | Feb 3, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8987443 | Mar 5, 2014 | Mar 24, 2015 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9023840 | Feb 21, 2014 | May 5, 2015 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US9034884 | Nov 18, 2011 | May 19, 2015 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| US9079912 | May 12, 2014 | Jul 14, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase inhibitors |
| US9193733 | May 17, 2013 | Nov 24, 2015 | Incyte Holdings Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US9206187 | Sep 6, 2013 | Dec 8, 2015 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase |
| US9216984 | Nov 8, 2013 | Dec 22, 2015 | Incyte Corporation | 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane—or heptane-nitrile as JAK inhibitors |
| US9221845 | Mar 11, 2015 | Dec 29, 2015 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |
PHOSHATE SEE……http://www.medchemexpress.com/product_pdf/HY-15315A/Baricitinib%20phosphate-NMR-HY-15315A-08874-2013.pdf
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 2-[1-ethylsulfonyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile | |
| CLINICAL DATA | |
| LEGAL STATUS |
|
| IDENTIFIERS | |
| CAS NUMBER | 1187594-09-7 |
| ATC CODE | None |
| PUBCHEM | CID: 44205240 |
| CHEMSPIDER | 26373084 |
| CHEMBL | CHEMBL2105759 |
| PDB LIGAND ID | 3JW (PDBe, RCSB PDB) |
| CHEMICAL DATA | |
| FORMULA | C16H17N7O2S |
| MOLECULAR MASS | 371.42 g/mol |
SEE..........http://newdrugapprovals.org/2013/06/17/lilly-and-partner-incyte-corp-have-presented-more-promising-data-on-their-investigational-jak-inhibitor-baricitinib-for-rheumatoid-arthritis/
//////////////
CCS(=O)(=O)N1CC(C1)(CC#N)N2C=C(C=N2)C3=C4C=CNC4=NC=N3