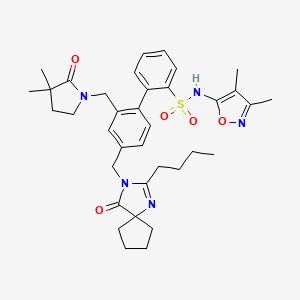

2-[4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-[(3,3-dimethyl-2-oxopyrrolidin-1-yl)methyl]phenyl]-N-(3,4-dimethyl-1,2-oxazol-5-yl)benzenesulfonamide
4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide,
4'- . (2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non-l-en-3-yl)methyll -N-C3.4- dimethyl-5-isoxazolyl)-2,-[(3.3-dimethyl-2-oxo-l- pyrrolidinvDmethyll [1.1 '-biphenyl] -2-sulfonamide
4‘-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide
BMS-248360
PRECLINICAL .....treating hypertension
Bristol Myers Squibb Co, INNOVATOR
Hypertension remains one of the largest unmet medical needs in the 21st century, especially when one considers that hypertension is the portent of future debilitating cardiovascular disease. While many drugs are available for treating the disease, approximately one-third of the hypertensive population is still not adequately treated. Of the more recent avenues explored for treating hypertension, disruption of the effects of either angiotensin II (AII) or endothelin-1 (ET-1) has shown promise. These endogenous vasoactive peptides are among the most potent vasoconstrictors and cell proliferative factors identified to date. AII is the effector molecule of the renin−angiotensin system (RAS), and a large number of AII receptor (AT1) antagonists, including irbesartan , have been developed for treating hypertension


SYNTHESIS
picked from.......http://www.drugfuture.com/synth/syndata.aspx?ID=324487

EP 1094816; JP 2002519380; US 2002143024; WO 0001389
The intermediate biphenyl aldehyde (XI) is prepared by two related methods. 4-Bromo-3-methylbenzonitrile (I) is oxidized to aldehyde (II) via radical bromination with N-bromosuccinimide/benzoyl peroxide, followed by treatment with trimethylamine N-oxide. Suzuki coupling of aryl bromide (II) with the pinacol boronate (III) affords biphenyl (IV). After protection of the aldehyde moiety of (IV) as the corresponding ethylene ketal (V), its cyano group is reduced to aldehyde (VI) employing DIBAL in THF. Subsequent reduction of (VI) with NaBH4 leads to alcohol (VII), which is further converted into the benzyl bromide (VIII) by means of CBr4/PPh3. Bromide (VIII) is condensed with the spiro imidazolone (IX) in the presence of NaH, to produce (X). Then acidic hydrolysis of the ethylene ketal and SEM groups of (X) gives rise to the intermediate aldehyde (XI)
NEXT

Alternatively, reduction of 4-bromo-3-formylbenzonitrile ethylene ketal (XII) by means of DIBAL leads to aldehyde (XIII), which is further reduced to alcohol (XIV) with NaBH4. After bromination of (XIV) with CBr4/PPh3, the resultant benzyl bromide (XV) is condensed with the spiro imidazolone (IX), yielding (XVI). Then, acidic ketal hydrolysis in (XVI) furnishes aldehyde (XVII). Suzuki coupling between aryl bromide (XVII) and boronic acid (XVIII) gives biphenyl (XIX). The SEM group of (XIX) is then removed under acidic conditions to provide (XI)

Reductive amination of the biphenyl aldehyde (XI) with 4-amino-2,2-dimethylbutanoic acid (XX) in the presence of NaBH(OAc)3 produces aminoacid (XXI). This is finally cyclized to the corresponding lactam by treatment with DIC

Coupling of 2-bromobenzenesulfonyl chloride (I) with 5-amino-3,4-dimethylisoxazole (II) affords sulfonamide (III), which is further protected as the N-methoxyethoxymethyl derivative (IV) employing MEM-chloride in DMF. Lithiation of bromosulfonamide (IV), followed by treatment with trimethyl borate and acidic work up leads to the boronic acid intermediate (V). This is then subjected to Suzuki coupling with 4-bromo-3-methylbenzaldehyde (VI) to yield the biphenyl adduct (VII). After reduction of aldehyde (VII) to the benzylic alcohol (VIII) with NaBH4, reaction with methanesulfonyl chloride and diisopropylethylamine gives rise to the mesylate (IX) (1-3).

Mesylate (IX) is condensed with ethyl 2-propyl-4-ethylimidazole-5-carboxylate (X) yielding (XI). Simultaneous ester group hydrolysis and MEM group deprotection under acidic conditions gives rise to the imidazolecarboxylic acid (XII). This is finally coupled with methylamine via activation with CDI to produce the desired N-methyl carboxamide (1-3).
Reductive amination of the biphenyl aldehyde (XI) with 4-amino-2,2-dimethylbutanoic acid (XX) in the presence of NaBH(OAc)3 produces aminoacid (XXI). This is finally cyclized to the corresponding lactam by treatment with DIC
PAPER
J. Med. Chem., 2002, 45 (18), pp 3829–3835
DOI: 10.1021/jm020138n
 BMS 248360
BMS 248360
The ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
15 as a white solid (40 mg, 31%):
mp 104−110 °C;
1H NMR (CDCl3) δ 0.90 (t, J = 7.0 Hz, 3H), 1.08 (s, 3H), 1.14 (s, 3H), 1.36 (m, 2H), 1.61 (m, 2H), 1.75−2.06 (m, 13H), 2.17 (s, 3H), 2.39 (m, 2H), 4.18 (m, 2H), 4.71 (m, 2H), 7.02−7.93 (m, 7H);
13CNMR (CDCl3 ) δ 7.82, 11.91, 14.79, 23.36, 25.50, 25.61, 27.11, 28.81, 29.88, 35.33, 38.42, 41.48, 44.59, 46.24, 46.47, 109.29, 125.15, 125.76, 129.68, 130.58, 131.76, 133.20, 134.07, 137.15, 138.27, 139.11, 139.57, 155.81, 162.68, 162.91, 181.25, 187.83.
Anal. (C36H45N5O5S) C, H, N, S.
.................................
PATENT
US 2002143024
 Zhang, H.-Y. et al., Tetrahedron, 1994, 50, 11339-11362.
Zhang, H.-Y. et al., Tetrahedron, 1994, 50, 11339-11362.
N-(3,4-Dimethyl-5-iso-xazolyl)-2′-formyl-4′-(hydroxy-methyl)-N-[[2-(tri-methylsilyl)ethoxy]- methyl][1,1′- biphenyl]-2- sulfonamide
Example 3 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
[0414]

Example 3 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide


A. 4′-Cyano-2′-(1,3-dioxolan-2-yl)-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
A mixture of 2B (1.28 g, 2.73 mmol), ethylene glycol (1.69 g, 27.3 mmol) and p-toluenesulfonic acid (38 mg) in toluene (30 mL) was heated at 130° C. for 5 h, while a Dean-Stark water separator was used. After cooling, the mixture was diluted with EtOAc. The organic liquid was separated and washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 5:4 hexane/EtOAc to afford 3A (1.1 g, 79%) as a colorless gum: Rf=0.57, silica gel, 1:2 hexane/EtOAc.
B. 2′-(1,3-Dioxolan-2-yl)-4′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
To 3A (1.1 g, 2.14 mmol) in THF (21 mL) at 0° C. was added DIBAL-H (1M in CH2Cl2, 4.28 mL 4.28 mmol) dropwise. The reaction was stirred at RT overnight. MeOH (20 mL) was added and the reaction was stirred for 5 min. The mixture was poured into cold 0.1 N HCl solution (150 mL), shaken for 5 min, and then extracted with 3:1 EtOAc/hexane. The combined organic extracts were washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 3:4 hexane/EtOAc to afford 3B (710 mg, 64%) as a colorless gum: Rf=0.45, silica gel, 2:3 hexane/EtOAc.
C. 2′-(1,3-Dioxolan-2-yl)-4′-hydroxymethyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) [1,1′-biphenyl]-2-sulfonamide
3B (710 mg, 1.4 mmol) was subjected to sodium borohydride reduction according to General Method 11 to afford 3C, which was used for the next reaction step without further purification.
D. 4′-Bromomethyl-2′-(1,3-dioxolan-2-yl)-N-(3,4′-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) [1,1′-biphenyl]-2-sulfonamide
3C was treated with carbon tetrabromide and triphenylphosphine according to General Method 2. The crude residue was chromatographed on silica gel using 3:2 hexane/EtOAc to afford 3D (750 mg, 94%) as a colorless gum: Rf=0.74, silica gel, 1:2 hexane/EtOAc.
E. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-(1,3-dioxolan-2-yl)-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
3D (750 mg, 1.3 mmol) was treated with 2-n-butyl-1,3-diazaspiro[4.4]non-1-en-4-one hydrochloride (387 mg, 1.68 mmol) according to General Method 4. The crude residue was chromatographed on silica gel using 100:1.7 CH2Cl2/MeOH to afford 3E as a gum (830 mg, 93%): Rf=0.40, silica gel, 100:5 CH2Cl2/MeOH.
F. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
3E (830 mg, 1.20 mmol) was subjected to deprotection according to General Method 7. The crude residue was chromatographed on silica gel using 100:1.5 and then 100:4 CH2Cl2 /MeOH to afford the title compound as a gum (480 mg, 72%): Rf=0.16, silica gel, 100:5 CH2Cl2/MeOH.
Example 4 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2′-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl][1,1′-biphenyl]-2-sulfonamide
To 3F (110 mg, 0.20 mmol) in CH2Cl2 (4 mL) was added 4-amino-2,2-dimethylbutanoic acid hydrochloride (98 mg, 0.59 mmol) [Scheinmann, et al., J. Chem. Research (S), 414-415 (1993)] and 3 Å molecular sieves, followed by glacial acetic acid (35 mg, 0.59 mmol) and then sodium acetate (48 mg, 0.59 mmol). The mixture was stirred for 8 minutes, and NaB(AcO)3H (124 mg, 0.59 mmol) was then added. The reaction mixture was stirred at RT for 2 h, diluted with EtOAc and filtered through celite. The filtrate was washed with H2O and brine, dried and concentrated. This material was dissolved in CH2Cl2 (6 mL) and 1,3-diisopropylcarbodiimide (32 mg, 0.25 mmol) was added. The reaction mixture was stirred at RT for 2 h and diluted with CH2Cl2, washed with H2O and brine, dried and concentrated. The residue was purified by preparative HPLC to provide the title compound as a white solid (40 mg, 31%, for two steps): mp 104-110° C. Analysis calculated for C36H45N5O5S.0.8 H2O: Calc'd: C, 64.13; H, 6.97; N, 10.39; S, 4,75. Found: C, 64.18; H, 6.60; N, 10.23; S, 4.50.
Example 5 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide (Alternative Preparation for 3F)
A. 2-[(2′-Bromo-5′-formyl)phenyl)]-1,3-dioxolane
DIBAL-H (1.0 M solution in toluene, 445 mL, 445 mmol, 1.1 eq) was added over 30 minutes to a solution of 2-[(2′-bromo-5′-cyano)phenyl)]-1,3-dioxolane (103 g, 404 mmol, 1.0 eq) [Zhang, H.-Y. et al., Tetrahedron, 50, 11339-11362 (1994)] in toluene (2.0 L) at −78° C. The solution was allowed to warm to 0° C. After 1 hour, a solution of Rochelle's salt (125 g) in water (200 mL) was added, and the mixture was allowed to warm to room temperature and was stirred vigorously for 16 h. The organic layer was concentrated and the residue partitioned between ethyl acetate (1 L) and 1 N hydrochloric acid (800 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (800 mL), dried over sodium sulfate, and then concentrated to give 70.5 g of crude 5A as a yellow solid, which was used without further purification.
B. 2-[(2′-Bromo-5′-hydroxymethyl)phenyl)]-1,3-dioxolane
Sodium borohydride (3.66 g, 96.7 mmol, 0.5 eq) was added to a solution of crude 5A (49.7 g, approximately 193 mmol, 1.0 eq) in absolute ethanol (1300 mL) at 0° C. After 2 hours, a solution of 10% aqueous sodium dihydrogen phosphate (50 mL) was added and the mixture was stirred and allowed to warm to room temperature. The mixture was concentrated, then partitioned between ethyl acetate (800 mL) and saturated aqueous sodium bicarbonate (500 mL). The organic layer was dried over sodium sulfate and concentrated to give 49.0 g of crude 5B as a yellow oil, which was used without further purification.
C. 2-[(2′-Bromo-5′-bromomethyl)phenyl)]-1,3-dioxolane
Triphenylphosphine (52.7 g, 199 mmol, 1.05 eq) was added in portions over 15 minutes to a solution of crude 5B (49.0 g, approximately 189 mmol, 1.0 eq) and carbon tetrabromide (69.0 g, 208 mmol, 1.1 eq) in THF at 0° C. After 2 hours, saturated aqueous sodium bicarbonate solution (20 mL) was added, and the mixture was allowed to warm to room temperature and was then concentrated. Ether (500 mL) was added, and the resulting mixture was filtered. The filtrate was dried over magnesium sulfate and concentrated. The residue was chromatographed on silica gel (8:1 hexanes/ethyl acetate as eluant) to give 5C as a white solid (31.1 g, 51% yield from 2-[(2′-bromo-5′-cyano)phenyl)]-1,3-dioxolane).
D. 2-(1,3-Dioxolan-2-yl)-4-[(2-n-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]bromobenzene
[0436] Sodium hydride (60% dispersion in mineral oil, 9.65 g, 241 mmol, 2.5 eq) was added in portions over 15 minutes to a mixture of 2-n-butyl-1,3-diazaspiro[4.4]non-1-en-4-one hydrochloride (18.7 g, 96.5 mmol, 1.0 eq) in DMF (400 mL) at 0° C. The mixture was stirred and allowed to warm to room temperature over 15 minutes. To this mixture was added via canula a solution of 5C (31.1 g, 96.5 mmol, 1.0 eq) in DMF (100 mL). After 14 hours, the mixture was concentrated in vacuo and partitioned between ethyl acetate (500 mL) and 10% aqueous sodium dihydrogen phosphate (300 mL). The organic layer was dried over sodium sulfate and concentrated to give crude 5D as an orange oil (42.7 g), which was used without further purification.
E. 4-[(2-n-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-formyl-bromobenzene
A solution of crude 5D (6.0 g, approximately 13.6 mmol, 1.0 eq) in THF (180 mL) and 1N hydrochloric acid (30 mL) was heated at 65° C. for 1.5 hours. The mixture was cooled and then treated with saturated aqueous sodium carbonate solution (75 mL) and ethyl acetate (200 mL). The organic layer was removed and dried over sodium sulfate, concentrated, and then further dried azeotropically with toluene to give 5E as a crude yellow oil (8.2 g) which contained a small amount of toluene. This material was used without further purification.
F. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
Palladium catalyzed Suzuki coupling of 5E and [2-[[(3,4-dimethyl-5-isoxazolyl)[(2-methoxyethoxy)methyl]amino]sulfonyl]phenyl]boronic acid was performed according to General Method 1 to yield 5F in 60% yield.
G. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
Deprotection of 5F according to General Method 7 provided the title compound (5G=3F) in 73% yield: Rf=0.2 (silica gel using CH2Cl2/MeOH [100:5]).
PATENT
EP 1237888; WO 0144239
Example 3 4'-r(2-Butyl-4-oxo-1.3-diazaspiror4.41non-l-en-3-yl)methvn-2'-formyl-N-
(3, 4-dimethyl-5-isoxazolyl)-[ 1,1 '-biphenyl] -2-sulfonamide

A. 4'-Cvano-2>-(1.3-dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [1.1 '-biphenyl] -2-sulfonamide
A mixture of 2B (1.28 g, 2.73 mmol), ethylene glycol (1.69 g, 27.3 mmol) and p-toluenesulfonic acid (38 mg) in toluene (30 mL) was heated at 130°C for 5 h, while a Dean-Stark water separator was used. After cooling, the mixture was diluted with EtOAc. The organic liquid was separated and washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 5:4 hexane/EtOAc to afford 3A (1.1 g, 79%) as a colorless gum: R^0.57, silica gel, 1:2 hexane EtOAc.
B. 2,-(1.3-Dioxolan-2-yl)-4'-formyl-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [1 , l'-biphenyl] -2-sulfonamide To 3A (1.1 g, 2.14 mmol) in THF (21 mL) at 0°C was added DIBAL- H (IM in CH2C12, 4.28 mL 4.28 mmol) dropwise. The reaction was stirred at RT overnight. MeOH (20 mL) was added and the reaction was stirred for 5 min. The mixture was poured into cold 0.1 N HCI solution (150 mL), shaken for 5 min, and then extracted with 3:1 EtOAc/hexane. The combined organic extracts were washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 3:4 hexane/EtOAc to afford 3B (710 mg, 64%) as a colorless gum: R^O.45, silica gel, 2:3 hexane/EtOAc. C. 2'-(1.3-Dioxolan-2-yl)-4'-hvdroxymethyl-N-(3.4-dimethyl-5- isoxazolyl)-N-(2-methoxyethoxymethyl) [1.1 '-biphenyl] -2- sulfonamide
3B (710 mg, 1.4 mmol) was subjected to sodium borohydride reduction according to General Method 11 to afford 3C, which was used for the next reaction step without further purification.
D. 4l-Bromomethyl-2,-(1.3-dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)- N-(2-methoxyethoxymethyl) [1 , l'-biphenyl] -2-sulfonamide 3C was treated with carbon tetrabromide and triphenylphosphine according to General Method 2. The crude residue was chromatographed on silica gel using 3:2 hexane/EtOAc to afford 3D (750 mg, 94%) as a colorless gum: R^0.74, silica gel, 1:2 hexane/EtOAc.
E. 4'-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn- 2,-(1.3- dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [ 1. l'-biphenyll -2-sulfonamide 3D (750 mg, 1.3 mmol) was treated with 2-re-butyl-l,3- diazaspiro[4.4]non-l-en-4-one hydrochloride (387 mg, 1.68 mmol) according to General Method 4. The crude residue was chromatographed on silica gel using 100:1.7 CH2CL/MeOH to afford 3E as a gum (830 mg, 93%): R^O.40, silica gel, 100:5 CH2Cl2/MeOH.
F. 4'-r(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2,- formyl-N-(3.4-dimethyl-5-isoxazolyl)-[l.l'-biphenyl1-2-sulfonamide
3E (830 mg, 1.20 mmol) was subjected to deprotection according to General Method 7. The crude residue was chromatographed on silica gel using 100:1.5 and then 100:4 CH2C12 /MeOH to afford the title compound as a gum (480 mg, 72%): R^O.16, silica gel, 100:5 CH.Cl MeOH.
Example 4
4'- . (2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non-l-en-3-yl)methyll -N-C3.4- dimethyl-5-isoxazolyl)-2,-[(3.3-dimethyl-2-oxo-l- pyrrolidinvDmethyll [1.1 '-biphenyl] -2-sulfonamide
To 3F (110 mg, 0.20 mmol) in CH2C12 (4 mL) was added 4-amino- 2,2-dimethylbutanoic acid hydrochloride (98 mg, 0.59 mmol) [Scheinmann, et al., J. Chem. Research (S), 414-415 (1993)] and 3A molecular sieves, followed by glacial acetic acid (35 mg, 0.59 mmol) and then sodium acetate (48 mg, 0.59 mmol). The mixture was stirred for 8 minutes, and NaB(AcO)3H (124 mg, 0.59 mmol) was then added. The reaction mixture was stirred at RT for 2 h, diluted with EtOAc and filtered through celite. The filtrate was washed with H2O and brine, dried and concentrated. This material was dissolved in CH2C12 (6 mL) and 1,3-diisopropylcarbodiimide (32 mg, 0.25 mmol) was added. The reaction mixture was stirred at RT for 2 h and diluted with CH2C12, washed with H2O and brine, dried and concentrated. The residue was purified by preparative HPLC to provide the title compound as a white solid (40 mg, 31%, for two steps): mp 104- 110°C. Analysis calculated for C36H45N5O5S • 0.8 H2O: Calc'd: C, 64.13; H, 6.97; N, 10.39; S, 4,75. Found: C, 64.18; H, 6.60; N, 10.23; S, 4.50.
Example 5
4'-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2,-formyl-N-
(3,4-dimethyl-5-isoxazolyl)-[l,l'-biphenyl]-2-sulfonamide (Alternative
Preparation for 3F)
A. 2-[(2'-Bromo-5'-formyl)phenyl)1-1.3-dioxolane
DIBAL-H (1.0 M solution in toluene, 445 mL, 445 mmol, 1.1 eq) was added over 30 minutes to a solution of 2-[(2'-bromo-5'-cyano)phenyl)]-l,3- dioxolane (103 g, 404 mmol, 1.0 eq) [Zhang, H.-Y. et al., Tetrahedron, 50, 11339-11362 (1994)] in toluene (2.0 L) at -78 °C. The solution was allowed to warm to 0 °C. After 1 hour, a solution of Rochelle's salt (125 g) in water (200 mL) was added, and the mixture was allowed to warm to room temperature and was stirred vigorously for 16 h. The organic layer was concentrated and the residue partitioned between ethyl acetate (1 L) and 1 N hydrochloric acid (800 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (800 mL), dried over sodium sulfate, and then concentrated to give 70.5 g of crude 5A as a yellow solid, which was used without further purification.
B. 2-[(2'-Bromo-5'-hvdroxymethyl)phenyl)l-1.3-dioxolane
Sodium borohydride (3.66 g, 96.7 mmol, 0.5 eq) was added to a solution of crude 5A (49.7 g, approximately 193 mmol, 1.0 eq) in absolute ethanol (1300 mL) at 0 °C. After 2 hours, a solution of 10% aqueous sodium dihydrogen phosphate (50 mL) was added and the mixture was stirred and allowed to warm to room temperature. The mixture was concentrated, then partitioned between ethyl acetate (800 mL) and saturated aqueous sodium bicarbonate (500 mL). The organic layer was dried over sodium sulfate and concentrated to give 49.0 g of crude 5B as a yellow oil, which was used without further purification. C. 2-[(2'-Bromo-5'-bromomethyl)phenyl)]-l,3-dioxolane Triphenylphosphine (52.7 g, 199 mmol, 1.05 eq) was added in portions over 15 minutes to a solution of crude 5B (49.0 g, approximately 189 mmol, 1.0 eq) and carbon tetrabromide (69.0 g, 208 mmol, 1.1 eq) in THF at 0 °C. After 2 hours, saturated aqueous sodium bicarbonate solution (20 mL) was added, and the mixture was allowed to warm to room temperature and was then concentrated. Ether (500 mL) was added, and the resulting mixture was filtered. The filtrate was dried over magnesium sulfate and concentrated. The residue was chromatographed on silica gel (8:1 hexanes/ethyl acetate as eluant) to give 5C as a white solid (31.1 g, 51% yield from 2-[(2'-bromo-5'-cyano)phenyl)]-l,3-dioxolane).
D. 2-( 1 ,3-Dioxolan-2-yl)-4- [ (2-re-butyl-4-oxo- 1 ,3-diazaspiro [4.4] non- 1- en-3-yl)methyl] bromobenzene Sodium hydride (60% dispersion in mineral oil, 9.65 g, 241 mmol,
2.5 eq) was added in portions over 15 minutes to a mixture of 2-rc-butyl- l,3-diazaspiro[4.4]non-l-en-4-one hydrochloride (18.7 g, 96.5 mmol, 1.0 eq) in DMF (400 mL) at 0°C. The mixture was stirred and allowed to warm to room temperature over 15 minutes. To this mixture was added via canula a solution of 5C (31.1 g, 96.5 mmol, 1.0 eq) in DMF (100 mL). After 14 hours, the mixture was concentrated in vacuo and partitioned between ethyl acetate (500 mL) and 10% aqueous sodium dihydrogen phosphate (300 mL). The organic layer was dried over sodium sulfate and concentrated to give crude 5D as an orange oil (42.7 g), which was used without further purification.
E. 4-[(2-n-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2- formyl-bromobenzene
A solution of crude 5D (6.0 g, approximately 13.6 mmol, 1.0 eq) in THF (180 mL) and IN hydrochloric acid (30 mL) was heated at 65°C for 1.5 hours. The mixture was cooled and then treated with saturated aqueous sodium carbonate solution (75 mL) and ethyl acetate (200 mL). The organic layer was removed and dried over sodium sulfate, concentrated, and then further dried azeotropically with toluene to give 5E as a crude yellow oil (8.2 g) which contained a small amount of toluene. This material was used without further purification.
F. 4'-.(2-Butyl-4-oxo-1.3-diazaspiro■4.41non-l-en-3-yl)methyl1-2,- formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) f 1.1 '-biphenyl] -2-sulfonamide Palladium catalyzed Suzuki coupling of 5E and [2-[[(3,4-dimethyl-5- isoxazolyl) [(2-methoxyethoxy)methyl] amino] sulfonyl] phenyl]boronic acid was performed according to General Method 1 to yield 5F in 60% yield.
G. 4'-[ 2-Butyl-4-oxo-1.3-diazaspiro[4■41non-l-en-3-yl)methvn-2,- formyl-N-(3 ,4-dimethyl-5-isoxazolyl)- fi .1 '-biphenyl] -2-sulfonamide
Deprotection of 5F according to General Method 7 provided the title compound (5G = 3F) in 73% yield: R^0.2 (silica gel using CH2ClJ eOH [100:5]).
| Patent | Submitted | Granted |
|---|---|---|
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6638937] | 2002-10-03 | 2003-10-28 |
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6835741] | 2004-06-03 | 2004-12-28 |
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6852745] | 2004-07-01 | 2005-02-08 |
///////////BMS-248360, Preclinical, SARTAN, BMS, HYPERTENTION
CCCCC1=NC2(CCCC2)C(=O)N1CC3=CC(=C(C=C3)C4=CC=CC=C4S(=O)(=O)NC5=C(C(=NO5)C)C)CN6CCC(C6=O)(C)C