

Trelagliptin
865759-25-7; UNII-Q836OWG55H
| Molecular Formula: | C18H20FN5O2 |
|---|---|
| Molecular Weight: | 357.382103 g/mol |
2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxopyrimidin-1-yl]methyl]-4-fluorobenzonitrile
(R) -2 - ((6 (3-amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) - yl) methyl) synthesis of 4-fluoro-benzonitrile
(R)-2-((6-(3-amino-3-methylpiperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)methyl)-4-fluorobenzonitrile
A dipeptidyl peptidase-4 (DPP-4) inhibitor used to treat type 2 diabetes.
Research Code SYR-472
CAS No. 865759-25-7 (Trelagliptin)
CAS No. 865759-25-7 (Trelagliptin)
1029877-94-8 (Trelagliptin Succinate)

Dipeptidyl Peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) is a type π membrane protein that has been referred to in the literature by a wide a variety of names including DPP4, DP4, DAP-IV, FAPβ, adenosine deaminase complexing protein 2, adenosine deaminase binding protein (AD Abp), dipeptidyl aminopeptidase IV; Xaa-Pro-dipeptidyl-aminopeptidase; Gly-Pro naphthylamidase; postproline dipeptidyl aminopeptidase IV; lymphocyte antigen CD26; glycoprotein GPI lO; dipeptidyl peptidase IV; glycylproline aminopeptidase; glycylproline aminopeptidase; X-prolyl dipeptidyl aminopeptidase; pep X; leukocyte antigen CD26; glycylprolyl dipeptidylaminopeptidase; dipeptidyl-peptide hydrolase; glycylprolyl aminopeptidase; dipeptidyl-aminopeptidase IV; DPP ΓV/CD26; amino acyl-prolyl dipeptidyl aminopeptidase; T cell triggering molecule TρlO3; X-PDAP. Dipeptidyl Peptidase IV is referred to herein as "DPP-IV." [0003] DPP-W is a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV dependent slow release of dipeptides of the type X-GIy or X-Ser has also been reported for some naturally occurring peptides.
DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of disease states, some of which are discussed below.
[0005] DPP-IV is responsible for the metabolic cleavage of certain endogenous peptides (GLP-I (7-36), glucagon) in vivo and has demonstrated proteolytic activity against a variety of other peptides (GHRH, NPY, GLP-2, VIP) in vitro.
DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of disease states, some of which are discussed below.
[0005] DPP-IV is responsible for the metabolic cleavage of certain endogenous peptides (GLP-I (7-36), glucagon) in vivo and has demonstrated proteolytic activity against a variety of other peptides (GHRH, NPY, GLP-2, VIP) in vitro.
GLP-I (7-36) is a 29 amino-acid peptide derived by post-translational processing of proglucagon in the small intestine. GLP-I (7-36) has multiple actions in vivo including the stimulation of insulin secretion, inhibition of glucagon secretion, the promotion of satiety, and the slowing of gastric emptying. Based on its physiological profile, the actions of GLP-I (7-36) are believed to be beneficial in the prevention and treatment of type II diabetes and potentially obesity. For example, exogenous administration of GLP-I (7-36) (continuous infusion) in diabetic patients has been found to be efficacious in this patient population. Unfortunately, GLP-I (7-36) is degraded rapidly in vivo and has been shown to have a short half -life in vivo (t1/2=1.5 minutes).
Based on a study of genetically bred DPP-IV knock out mice and on in vivo I in vitro studies with selective DPP-IV inhibitors, DPP-IV has been shown to be the primary degrading enzyme of GLP-I (7-36) in vivo. GLP-I (7-36) is degraded by DPP-IV efficiently to GLP-I (9-36), which has been speculated to act as a physiological antagonist to GLP-I (7-36). Inhibiting DPP-TV in vivo is therefore believed to be useful for potentiating endogenous levels of GLP-I (7-36) and attenuating the formation of its antagonist GLP-I (9-36). Thus, DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV, in particular diabetes and more particularly, type 2 diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (WG), metabolic acidosis, ketosis, appetite regulation and obesity.
Based on a study of genetically bred DPP-IV knock out mice and on in vivo I in vitro studies with selective DPP-IV inhibitors, DPP-IV has been shown to be the primary degrading enzyme of GLP-I (7-36) in vivo. GLP-I (7-36) is degraded by DPP-IV efficiently to GLP-I (9-36), which has been speculated to act as a physiological antagonist to GLP-I (7-36). Inhibiting DPP-TV in vivo is therefore believed to be useful for potentiating endogenous levels of GLP-I (7-36) and attenuating the formation of its antagonist GLP-I (9-36). Thus, DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV, in particular diabetes and more particularly, type 2 diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (WG), metabolic acidosis, ketosis, appetite regulation and obesity.
DPP-IV expression is increased in T-cells upon mitogenic or antigenic stimulation (Mattem, T., et al., Scand. J. Immunol, 1991, 33, 737). It has been reported that inhibitors of DPP-IV and antibodies to DPP-IV suppress the proliferation of mitogen-stimulated and antigen-stimulated T-cells in a dose-dependant manner (Schon, E., et al., Biol. Chem., 1991, 372, 305). Various other functions of T-lymphocytes such as cytokine production, IL-2 mediated cell proliferation and B-cell helper activity have been shown to be dependent on DPP-IV activity (Schon, E., et al., Scand. J. Immunol, 1989, 29, 127). DPP-IV inhibitors, based on boroProline, (Flentke, G. R., et al., Proc. Nat. Acad. Set USA, 1991, 88, 1556) although unstable, were effective at inhibiting antigen-induced lymphocyte proliferation and IL-2 production in murine CD4+ T-helper cells. Such boronic acid inhibitors have been shown to have an effect in vivo in mice causing suppression of antibody production induced by immune challenge (Kubota, T. et al, Clin. Exp. Immun., 1992, 89, 192). The role of DPP-IV in regulating T lymphocyte activation may also be attributed, in part, to its cell-surface association with the transmembrane phosphatase, CD45. DPP-IV inhibitors or non-active site ligands may possibly disrupt the CD45-DPP-TV association. CD45 is known to be an integral component of the T-cell signaling apparatus. It has been reported that DPP-IV is essential for the penetration and infectivity of HTV-I and HTV-2 viruses in CD4+ T-cells (Wakselman, M., Nguyen, C, Mazaleyrat, J.-P., Callebaut, C, Krust, B., Hovanessian, A. G., Inhibition of HIV-I infection of CD 26+ but not CD 26-cells by a potent cyclopeptidic inhibitor of the DPP-IV activity of CD 26. Abstract P.44 of the 24.sup.th European Peptide Symposium 1996). Additionally, DPP-IV has been shown to associate with the enzyme adenosine deaminase (ADA) on the surface of T-cells (Kameoka, J., et al., Science, 193, 26 466). ADA deficiency causes severe combined immunodeficiency disease (SCID) in humans. This ADA-CD26 interaction may provide clues to the pathophysiology of SCID. It follows that inhibitors of DPP-TV may be useful immunosuppressants (or cytokine release suppressant drugs) for the treatment of among other things: organ transplant rejection; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; and the treatment of AIDS.
It has been shown that lung endothelial cell DPP-IV is an adhesion molecule for lung-metastatic rat breast and prostate carcinoma cells (Johnson, R. C, et al., J. Cell. Biol, 1993, 121, 1423). DPP-IV is known to bind to fibronectin and some metastatic tumor cells are known to carry large amounts of fibronectin on their surface. Potent DPP-IV inhibitors may be useful as drugs to prevent metastases of, for example, breast and prostrate tumors to the lungs.
[0010] High levels of DPP-PV expression have also been found in human skin fibroblast cells from patients with psoriasis, rheumatoid arthritis (RA) and lichen planus (Raynaud, F., et al., J. Cell. Physiol, 1992, 151, 378). Therefore, DPP-TV inhibitors may be useful as agents to treat dermatological diseases such as psoriasis and lichen planus. [0011] High DPP-TV activity has been found in tissue homogenates from patients with benign prostate hypertrophy and in prostatosomes. These are prostate derived organelles important for the enhancement of sperm forward motility (Vanhoof, G., et al., EMr. /.
It has been shown that lung endothelial cell DPP-IV is an adhesion molecule for lung-metastatic rat breast and prostate carcinoma cells (Johnson, R. C, et al., J. Cell. Biol, 1993, 121, 1423). DPP-IV is known to bind to fibronectin and some metastatic tumor cells are known to carry large amounts of fibronectin on their surface. Potent DPP-IV inhibitors may be useful as drugs to prevent metastases of, for example, breast and prostrate tumors to the lungs.
[0010] High levels of DPP-PV expression have also been found in human skin fibroblast cells from patients with psoriasis, rheumatoid arthritis (RA) and lichen planus (Raynaud, F., et al., J. Cell. Physiol, 1992, 151, 378). Therefore, DPP-TV inhibitors may be useful as agents to treat dermatological diseases such as psoriasis and lichen planus. [0011] High DPP-TV activity has been found in tissue homogenates from patients with benign prostate hypertrophy and in prostatosomes. These are prostate derived organelles important for the enhancement of sperm forward motility (Vanhoof, G., et al., EMr. /.
Clin. Chem. Clin. Biochem., 1992, 30, 333). DPP-IV inhibitors may also act to suppress sperm motility and therefore act as a male contraceptive agent. Conversely, DPP-IV inhibitors have been implicated as novel for treatment of infertility, and particularly human female infertility due to Polycystic ovary syndrome (PCOS, Stein-Leventhal syndrome) which is a condition characterized by thickening of the ovarian capsule and . formation of multiple follicular cysts. It results in infertility and amenorrhea.
DPP-IV is thought to play a role in the cleavage of various cytokines
(stimulating hematopoietic cells), growth factors and neuropeptides.
Stimulated hematopoietic cells are useful for the treatment of disorders that are characterized by a reduced number of hematopoietic cells or their precursors in vivo. Such conditions occur frequently in patients who are immunosuppressed, for example, as a consequence of chemotherapy and/or radiation therapy for cancer. It was discovered that inhibitors of dipeptidyl peptidase type PV are useful for stimulating the growth and differentiation of hematopoietic cells in the absence of exogenously added cytokines or other growth factors or stromal cells. This discovery contradicts the dogma in the field of hematopoietic cell stimulation, which provides that the addition of cytokines or cells that produce cytokines (stromal cells) is an essential element for maintaining and stimulating the growth and differentiation of hematopoietic cells in culture. (See, e.g., PCT Intl. Application No. PCT/US93/017173 published as WO 94/03055).
DPP-IV in human plasma has been shown to cleave N-terminal Tyr-Ala from growth hormone-releasing factor and cause inactivation of this hormone. Therefore, inhibitors of DPP-IV may be useful in the treatment of short stature due to growth hormone deficiency (Dwarfism) and for promoting GH-dependent tissue growth or re-growth.
DPP-IV can also cleave neuropeptides and has been shown to modulate the activity of neuroactive peptides substance P, neuropeptide Y and CLIP (Mentlein, R., Dahms, P., Grandt, D., Kruger, R., Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV, Regul. Pept., 49, 133, 1993; Wetzel, W., Wagner, T., Vogel, D., Demuth, H.-U., Balschun, D., Effects of the CLIP fragment ACTH 20-24 on the duration of REM sleep episodes, Neuropeptides, 31, 41, 1997). Thus DPP-IV inhibitors may also be useful agents for the regulation or normalization of neurological disorders.
Several compounds have been shown to inhibit DPP-IV. Nonetheless, a need still exists for new DPP-IV inhibitors that have advantageous potency, stability, selectivity, toxicity and/or pharmacodynamics properties. In this regard, synthetic methods are provided that can be used to make a novel class of DPP-IV inhibitors.
DPP-IV is thought to play a role in the cleavage of various cytokines
(stimulating hematopoietic cells), growth factors and neuropeptides.
Stimulated hematopoietic cells are useful for the treatment of disorders that are characterized by a reduced number of hematopoietic cells or their precursors in vivo. Such conditions occur frequently in patients who are immunosuppressed, for example, as a consequence of chemotherapy and/or radiation therapy for cancer. It was discovered that inhibitors of dipeptidyl peptidase type PV are useful for stimulating the growth and differentiation of hematopoietic cells in the absence of exogenously added cytokines or other growth factors or stromal cells. This discovery contradicts the dogma in the field of hematopoietic cell stimulation, which provides that the addition of cytokines or cells that produce cytokines (stromal cells) is an essential element for maintaining and stimulating the growth and differentiation of hematopoietic cells in culture. (See, e.g., PCT Intl. Application No. PCT/US93/017173 published as WO 94/03055).
DPP-IV in human plasma has been shown to cleave N-terminal Tyr-Ala from growth hormone-releasing factor and cause inactivation of this hormone. Therefore, inhibitors of DPP-IV may be useful in the treatment of short stature due to growth hormone deficiency (Dwarfism) and for promoting GH-dependent tissue growth or re-growth.
DPP-IV can also cleave neuropeptides and has been shown to modulate the activity of neuroactive peptides substance P, neuropeptide Y and CLIP (Mentlein, R., Dahms, P., Grandt, D., Kruger, R., Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV, Regul. Pept., 49, 133, 1993; Wetzel, W., Wagner, T., Vogel, D., Demuth, H.-U., Balschun, D., Effects of the CLIP fragment ACTH 20-24 on the duration of REM sleep episodes, Neuropeptides, 31, 41, 1997). Thus DPP-IV inhibitors may also be useful agents for the regulation or normalization of neurological disorders.
Several compounds have been shown to inhibit DPP-IV. Nonetheless, a need still exists for new DPP-IV inhibitors that have advantageous potency, stability, selectivity, toxicity and/or pharmacodynamics properties. In this regard, synthetic methods are provided that can be used to make a novel class of DPP-IV inhibitors.
Trelagliptin (Zafatek) is a pharmaceutical drug used for the treatment of type 2 diabetes (diabetes mellitus).[1]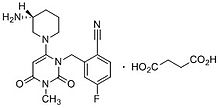
Indications for Medical Use
It is a highly selective dipeptidyl peptidase (DPP-4) inhibitor that is typically used as an add on treatment when the first line treatment of metformin is not achieving the expected glycemic goals; though it has been approved for use as a first line treatment when metformin cannot be used.[1]
Biochemistry
DPP-4 inhibitors activate T-cells and are more commonly known as T-cell activation antigens (specifically CD26).[1][2] Chemically, it is a fluorinated derivative of alogliptin.
Development
Formulated as the salt trelagliptin succinate, it was approved for use in Japan in March 2015.[3] Takeda, the company that developed trelagliptin, chose to not get approval for the drug in the USA and EU.[1] The licensing rights that Takeda purchased from Furiex Pharmaceuticals for DPP-4 inhibitors included a clause specific to development of this drug in the USA and EU.[1] The clause required that all services done for phase II and phase III clinical studies in the USA and EU be purchased through Furiex.[1] Takeda chose to cease development of this drug in the USA and EU because of the high costs quoted by Furiex for these services.[1] Gliptins have been on the market since 2006 and there are 8 gliptins currently registered as drugs (worldwide).[4] Gliptins are an emerging market and are thus being developed at an increasing rate; there are currently two gliptins in advanced stages of development that are expected to be on the market in the coming year.[4]
Gliptins are thought to have cardiovascular protective abilities though the extent of these effects is still being studied.[4] They are also being studied for the ability that this class of drugs has at promoting B-cell survival.[4]
Administration and Dosing
Similar drugs in the same class as trelagliptin are administered once daily while trelagliptin is administered once weekly.[1][5] Alogliptin (Nesina) is the other major DPP-4 inhibitor on the market. It is also owned by Takeda and is administered once daily. A dosing of once per week is advantageous as a reduction in the frequency of required dosing is known to increase patient compliance.[1][2]
Zafatek is administered in the form trelagliptin succinate in a 1:1 mixture of trelagliptin and succinic acid.[6] The drug is marketed with the IUPAC name Succinic acid - 2-({6-[(3R)-3-amino-1-piperidinyl]-3-methyl-2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl}methyl)-4-fluorobenzonitrile (1:1), has a molecular mass of 475.470143 grams/mol, and has the molecular formula | C=22 | H=26 | F=1 | N=5 | O=6 .[6][7]
SYNTHESIS ................
PAPER
J. Med .Chem.,2011, 54, 510-524
Synthesis started with selective alkylation of chlorouracil 80, followed by methylation provided compound153via152.
The displacement of chloride with 3-(R)-aminopiperidine83afforded trelagliptin154..


The discovery of two classes of heterocyclic dipeptidyl peptidase IV (DPP-4) inhibitors, pyrimidinones and pyrimidinediones, is described. After a single oral dose, these potent, selective, and noncovalent inhibitors provide sustained reduction of plasma DPP-4 activity and lowering of blood glucose in animal models of diabetes. Compounds 13a, 27b, and 27j were selected for development.
2-[6-(3-Aminopiperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluorobenzonitrile, TFA salt (27j)
A mixture of 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol), and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60 °C for 2 h. The mixture was diluted with water and extracted with EtOAc. The organics were dried over MgSO4, and the solvent was removed. The residue was purified by column chromatography to give 0.66 g of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluorobenzonitrile (60%). 1H NMR (400 MHz, CDCl3): δ 7.73 (dd, J = 7.2, 8.4 Hz, 1H), 7.26 (d, J = 4.0 Hz, 1H), 7.11−7.17 (m, 1H), 6.94 (dd, J = 2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [M + H] calcd for C13H9ClFN3O2, 293; found 293.
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluorobenzonitrile (300 mg, 1.0 mmol), 3-(R)-aminopiperidine dihydrochloride (266 mg, 1.5 mmol), and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 °C for 2 h. The final compound (367 mg, 81% yield) was obtained as a TFA salt after HPLC purification. 1H NMR (400 MHz, CD3OD): δ 7.77−7.84 (m, 1H), 7.16−7.27 (m, 2H), 5.46 (s, 1H), 5.17−5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33−3.47 (m, 2H), 3.22 (s, 3H), 2.98−3.08 (m, 1H), 2.67−2.92 (m, 2H), 2.07−2.17 (m, 1H), 1.82−1.92 (m, 1H), 1.51−1.79 (m, 2H). MS (ES) [M + H] calcd for C18H20FN5O2, 357; found, 357.
PATENT
WO 2007035629
(R)-2-((6-(3-amino-3-methylpiperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)methyl)-4-fluorobenzonitrile (30). 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-3-methyl-piperidine dihydrochloride (266 mg, 1.4 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 1000C for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.78-7.83 (m, IH), 7.14-7.26 (m, 2H), 5.47 (s, IH), 5.12-5.36 (ABq, 2H, J = 105.2, 15.6 Hz), 3.21 (s, IH), 2.72-3.15 (m, 4H), 1.75-1.95 (m, 4H), 1.39 (s, 3H). MS (ES) [m+H] calc'd for C19H22FN5O2, 372.41; found, 372.41.
Compound 34
Compound 34

4-Fluoro-2-methylbenzonitrile (31). A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32). A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
Alternatively, 32 was made as follows. 4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AJJBN (122 g) and heated to 750C. A suspension of DBH (353 g) in DCE (500 mL) was added at 750C portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AJ-BN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10°C over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20°C over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro-benzonitrile (33). A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 6O0C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=1.2, 8.4Hz, IH), 7.26 (d, J-4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc'd for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows. To a solution of 6-chloro-3-methyluracil (750 g) and W,iV-diisopropylethylarnine (998 mL) in NMP (3 L) was added (at <30°C over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 6O0C for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5°C for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2 X 2.25 L). The material was then dried in a vacuum oven at 40±5°C for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile (34). 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 1000C for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc'd for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 6O0C until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 6O0C and the mixture was allowed to cool to <25°C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2 X 3.6 L). The filtrate was concentrated at 450C under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 00C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc'd for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35°C over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 5O0C (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purifed water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 3O0C over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 6O0C until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57°C. After stirring at 6O0C for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5°C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2 X 4.2 L). The material was then dried in a vacuum oven at 70±5°C (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75°C and filtered through #3 Whatman filter paper. The solution was heated back to 750C and a IM solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20°C/hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
2-Bromomethyl-4-fluorobenzonitrile (32). A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
Alternatively, 32 was made as follows. 4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AJJBN (122 g) and heated to 750C. A suspension of DBH (353 g) in DCE (500 mL) was added at 750C portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AJ-BN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10°C over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20°C over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro-benzonitrile (33). A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 6O0C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=1.2, 8.4Hz, IH), 7.26 (d, J-4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc'd for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows. To a solution of 6-chloro-3-methyluracil (750 g) and W,iV-diisopropylethylarnine (998 mL) in NMP (3 L) was added (at <30°C over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 6O0C for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5°C for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2 X 2.25 L). The material was then dried in a vacuum oven at 40±5°C for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile (34). 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 1000C for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc'd for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 6O0C until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 6O0C and the mixture was allowed to cool to <25°C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2 X 3.6 L). The filtrate was concentrated at 450C under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 00C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc'd for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35°C over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 5O0C (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purifed water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 3O0C over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 6O0C until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57°C. After stirring at 6O0C for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5°C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2 X 4.2 L). The material was then dried in a vacuum oven at 70±5°C (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75°C and filtered through #3 Whatman filter paper. The solution was heated back to 750C and a IM solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20°C/hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
PATENT
US 2008227798
- EXAMPLES
- Compound I may be prepared by the follow synthetic route (Scheme 1)
- Example 1Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)


A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)
- Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).

B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)
- Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).

C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)
- Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc'd for C13H9ClFN3O2, 293.68; found 293.68.

D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)
- Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc'd for C18H20FN5O2, 357.38; found, 357.38.

E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile
- The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc'd for C18H20FN5O2, 357.38; found, 357.38.
- Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.

| TABLE A | |||
| Approximate Solubilities of Compound I | |||
| Solubility | |||
| Solvent | (mg/mL)a | ||
| Acetone | 2 | ||
| Acetonitrile (ACN) | <1 | ||
| Dichloromethane (DCM) | <1 | ||
| Dimethyl Formamide (DMF) | 68 | ||
| 1,4-Dioxane | <1 | ||
| Ethanol (EtOH) | 2 | ||
| Ethyl Acetate (EtOAc) | <1 | ||
| di-Ethyl ether | <1 | ||
| Hexanes | <1 | ||
| 2-Propanol (IPA) | <1 | ||
| Methanol (MeOH) | 20 | ||
| Tetrahydrofuran (THF) | <1 | ||
| Toluene | <1 | ||
| Trifluoroethanol (TFE) | >200 | ||
| Water (H2O) | 51 | ||
| ACN:H2O (85:15) | 101 | ||
| EtOH:H2O (95:5) | 5 | ||
| IPA:H2O (88:12) | 11 | ||
| aApproximate solubilities are calculated based on the total solvent used to give a solution; actual solubilities may be greater because of the volume of the solvent portions utilized or a slow rate of dissolution. Solubilities are reported to the nearest mg/mL. | |||
PATENT
Reference Example 2
in the following formula 2, 2 - ((6 - ((3R) -3- amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H ) - yl) shown in the following example of a production process of a methyl) -4-fluoro-benzonitrile succinate (4b).
in the following formula 2, 2 - ((6 - ((3R) -3- amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H ) - yl) shown in the following example of a production process of a methyl) -4-fluoro-benzonitrile succinate (4b).
[Formula 2]

[In the formula 2, 2 - ((6-chloro-3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) - yl) methyl) -4-fluorobenzonitrile (2b) manufacturing process]
ethyl acetate (3.5 vol), 2- (bromomethyl) -4-fluorobenzonitrile (1b) (1 equiv, 1wt.), 6- chloro-3-methyl uracil (1.05 eq, 0.79wt), N- methylpyrrolidone (NMP;.. 3.5 times the amount), diisopropylethylamine (Hunig's base, 2.1 eq, 1.27wt) was heated to an internal temperature of 60 ~ 70 ℃ a.
The mixture was stirred until 2-4 hours or the completion of the reaction at 60 ~ 70 ℃.
Then cooling the solution to 40 ~ 50 ℃, after stirring at least 30 minutes, 40 ~ 50 ℃ isopropanol (1.5 times) while maintaining, water (3.5 times the amount) was added, then at least one hour stirring did. The solution was cooled to 20 ~ 30 ℃, was then stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, was then stirred for at least 1 hour. The resulting slurry was filtered, washed with 0 ~ 10 ℃ in cold isopropanol (4.0 vol), and vacuum dried at 45 ~ 55 ℃, to give the above compound (2b).
ethyl acetate (3.5 vol), 2- (bromomethyl) -4-fluorobenzonitrile (1b) (1 equiv, 1wt.), 6- chloro-3-methyl uracil (1.05 eq, 0.79wt), N- methylpyrrolidone (NMP;.. 3.5 times the amount), diisopropylethylamine (Hunig's base, 2.1 eq, 1.27wt) was heated to an internal temperature of 60 ~ 70 ℃ a.
The mixture was stirred until 2-4 hours or the completion of the reaction at 60 ~ 70 ℃.
Then cooling the solution to 40 ~ 50 ℃, after stirring at least 30 minutes, 40 ~ 50 ℃ isopropanol (1.5 times) while maintaining, water (3.5 times the amount) was added, then at least one hour stirring did. The solution was cooled to 20 ~ 30 ℃, was then stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, was then stirred for at least 1 hour. The resulting slurry was filtered, washed with 0 ~ 10 ℃ in cold isopropanol (4.0 vol), and vacuum dried at 45 ~ 55 ℃, to give the above compound (2b).
[In the formula 2, 2 - ((6 - ((3R) -3- amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) - yl) methyl) -4-manufacturing process of the fluorobenzonitrile (3b)]
the above compound (2b) (1 eq, 1wt.), (R) -3- aminopiperidine dihydrochloride (1.1 eq, 0.65wt .), potassium carbonate (2.5 equivalents, 1.18wt.), isopropanol (5.0 vol), water (1.5 times) until the completion of the reaction with 65 ~ 75 ℃ (eg, 3 to 7 hours ) was allowed to react. Potassium carbonate in 65 ~ 75 ℃ (7.05 eq, 3.32wt.), Water (5.5 vol) was added, and after stirring for about 30 minutes, the phases were separated at 50 ℃ ~ 70 ℃. The organic solvent was concentrated under reduced pressure to approximately 5 times. And water (5 vol) was added to the solution and concentrated under reduced pressure to approximately 5 times. The solution was stirred for about 40 minutes at 55 ℃ ~ 75 ℃. The solution was cooled to 20 ℃ ~ 30 ℃, was then stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, subsequently stirred for at least 1 hour, the resulting slurry was filtered, washed with 0 ~ 10 ℃ in cold water (2.0 times the amount), 45 ~ 55 ℃ was vacuum dried to give the above compound (3b).
the above compound (2b) (1 eq, 1wt.), (R) -3- aminopiperidine dihydrochloride (1.1 eq, 0.65wt .), potassium carbonate (2.5 equivalents, 1.18wt.), isopropanol (5.0 vol), water (1.5 times) until the completion of the reaction with 65 ~ 75 ℃ (eg, 3 to 7 hours ) was allowed to react. Potassium carbonate in 65 ~ 75 ℃ (7.05 eq, 3.32wt.), Water (5.5 vol) was added, and after stirring for about 30 minutes, the phases were separated at 50 ℃ ~ 70 ℃. The organic solvent was concentrated under reduced pressure to approximately 5 times. And water (5 vol) was added to the solution and concentrated under reduced pressure to approximately 5 times. The solution was stirred for about 40 minutes at 55 ℃ ~ 75 ℃. The solution was cooled to 20 ℃ ~ 30 ℃, was then stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, subsequently stirred for at least 1 hour, the resulting slurry was filtered, washed with 0 ~ 10 ℃ in cold water (2.0 times the amount), 45 ~ 55 ℃ was vacuum dried to give the above compound (3b).
[In the above formula 2, the compound production step of succinate (4b) of (3b)]
Compound (3b), tetrahydrofuran (6.0 vol), isopropanol (3.0 vol), water (0. a 6-fold amount) was heated to 55 ~ 65 ℃. Tetrahydrofuran solution of succinic acid (20 ℃ ~ 30 ℃) was added and the solution was stirred for about 15 minutes and maintained at 55 ~ 65 ℃.
The solution was cooled to 20 ~ 30 ℃, the mixture was stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, was then stirred for at least 1 hour. After the resulting slurry filtered and washed with isopropanol (6.0 vol). The resulting wet crystals were dried at 65 ~ 75 ℃, was obtained succinate of the compound (3b) and (4b) as a white crystalline solid.
Compound (3b), tetrahydrofuran (6.0 vol), isopropanol (3.0 vol), water (0. a 6-fold amount) was heated to 55 ~ 65 ℃. Tetrahydrofuran solution of succinic acid (20 ℃ ~ 30 ℃) was added and the solution was stirred for about 15 minutes and maintained at 55 ~ 65 ℃.
The solution was cooled to 20 ~ 30 ℃, the mixture was stirred for at least 1 hour. The solution was cooled to 0 ~ 10 ℃, was then stirred for at least 1 hour. After the resulting slurry filtered and washed with isopropanol (6.0 vol). The resulting wet crystals were dried at 65 ~ 75 ℃, was obtained succinate of the compound (3b) and (4b) as a white crystalline solid.


PATENT
2 - ({6 -! [(3R) -3- amino-piperidin-1-yl] -3-methyl-dihydro-pyrimidin _3,4_ _2,4_ dioxo-1 (2 1) - yl} methyl) benzonitrile is an effective DPP-1V inhibitors class of drugs in recent years in Japan, the structural formula
As shown below.

Chinese Patent Application CN1926128 discloses a process for preparing 2_ ({6_ [(3R) -3- amino-piperidin-1-yl] -3-methyl-2,4-dioxo-3,4- dihydropyrimidine-1 (2 1!) - yl} methyl) benzonitrile method, as shown in Scheme I:

Scheme I
In the above reaction scheme, 6-chloro-uracil and 2-bromomethyl-benzene cyanide in a mixed solvent of DMF-DMSO, in the presence of NaH and LiBr alkylation reaction to give compound 2 in a yield of 54%. Compound 2 is further alkylation reaction of compound yield 3 is 72%. The total yield of the compound 4 prepared in 20% yield is low, and the preparation of compound 4 obtained purity is not high, but also the need for further purification, such as recrystallization, column chromatography and other means in order to obtain high-purity suitable Pharmaceutically acceptable 2 - ({6 - [(3R) -3- amino-piperidin-1-yl] -3-methyl-2,4-dioxo-3,4-dihydro-pyrimidin _1 (2! 1) - yl} methyl) benzonitrile compound. Preparation still find more suitable for industrial production, a higher yield of the 2- ({6- [(3R) -3- amino-piperidin-1-yl] -3-methyl-2,4-dioxo -3, (2Η) 4- dihydropyrimidine-1 - yl} methyl) benzonitrile or a salt or the like.


PATENT
Example 15
(R) -2 - ((6 (3-amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) - yl) methyl) synthesis of 4-fluoro-benzonitrile
(R) -2 - ((6 (3-amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) - yl) methyl) synthesis of 4-fluoro-benzonitrile

100mL four-necked flask of water and isopropanol 1/1 (v / v) mixture 60mL was added, pyridine 21.4μL [d = 0.98, mw.79.10, 0.26mmol], (R) -1- (3- (2 - cyano-5-fluoro-benzyl) -1-methyl-2,6-dioxo-1,2,3,6-tetra-hydro-4-yl) piperidin-3-carboxamide 2.00g [mw.385.39, 5.19mmol] of It was added to the order. Then, iodobenzene diacetate 1.84g [mw.322.10, 5.71mmol] was added, and the mixture was stirred for 3 h at 20 ℃. After volatile components were distilled off under reduced pressure by an evaporator, and the aqueous solution was washed twice with ethyl acetate 20mL. After cooling to near 0 ℃, potassium carbonate 16g added stepwise at 15 ℃ or less, was extracted by the addition of toluene 6mL and isopropanol 6mL. After separation, the organic layer was washed with saturated brine 10mL, adding toluene 6mL after concentration under reduced pressure by an evaporator, and further subjected to vacuum concentration. It was suspended by the addition of toluene 6mL to concentrate, by the addition of n-heptane 6mL, after 1 hour and aged at 0 ℃, reduced pressure filtration, to obtain the desired compound after drying under reduced pressure at 50 ℃. White crystalline powder, 1.6g, 86% yield.
1 H-NMR (500 MHz, CDCl 3 ) delta (ppm) 1.23 (D, J = 11.03 Hz, 1H) 1.30 (BRS, 2H) 1.56-1.67 (M, 1H) 1.72-1.83 (M, 1H) 1.95 (dd , J = 12.77 Hz, 3.94 Hz, 1H) 2.41 (m, 1H) 2.61 (m, 1H) 2.87-2.98 (m, 2H) 2.99-3.05 (m, 1H) 3.32 (s, 3H) 5.23-5.32 (m , 2H) 5.39 (s, 1H) 6.86 (dd, J = 8.99 Hz, 2.36 Hz, 1H) 7.09 (td, J = 8.04 Hz, 2.52 Hz, 1H) 7.69 (dd, J = 8.51 Hz, 5.36 Hz, 1H ).
13 C NMR (126 MHz, CDCl 3 ) ppm 28.0, 33.4, 46.1, 51.9, 59.7, 90.8, 114.6,114.7, 115.6, 115.8, 116.4, 135.4, 135.5, 144.6, 152.7, 159.5, 162.9.
Reference Example 4
(R) -2 - ((6 (3-amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) - yl) methyl) synthesis of 4-fluoro-benzonitrile succinate
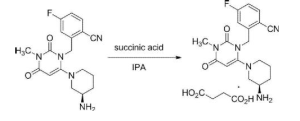
50mL eggplant-shaped flask (R) -2 - ((6- (3- amino-1-yl) -3-methyl-2,4-dioxo-3,4-dihydro-pyrimidine -1 (2H) - yl) methyl) -4-fluorobenzonitrile 1.0g [mw.357.38, 2.8mmol], it was added tetrahydrofuran 4.5mL and water 2 drops. After heated and dissolved at 65 ℃, was dropped to the solution was dissolved at the same temperature 0.331g succinic acid [mw.118.09, 2.8mmol] with tetrahydrofuran 4mL and isopropanol 2.5mL. Aged for 16 hours at room temperature after stirring for 30 min at 65 ℃, and stirred for a further 2 hours at 0 ℃. The crystallization product was collected by terrorism to vacuum filtration. To obtain the desired compound after drying under reduced pressure at 45 ℃. White crystalline powder, 1.2g, 93% yield.
Reference Example 4
(R) -2 - ((6 (3-amino-piperidin-1-yl) -3-methyl-2,4-dioxo-3,4-dihydropyrimidine -1 (2H) - yl) methyl) synthesis of 4-fluoro-benzonitrile succinate

50mL eggplant-shaped flask (R) -2 - ((6- (3- amino-1-yl) -3-methyl-2,4-dioxo-3,4-dihydro-pyrimidine -1 (2H) - yl) methyl) -4-fluorobenzonitrile 1.0g [mw.357.38, 2.8mmol], it was added tetrahydrofuran 4.5mL and water 2 drops. After heated and dissolved at 65 ℃, was dropped to the solution was dissolved at the same temperature 0.331g succinic acid [mw.118.09, 2.8mmol] with tetrahydrofuran 4mL and isopropanol 2.5mL. Aged for 16 hours at room temperature after stirring for 30 min at 65 ℃, and stirred for a further 2 hours at 0 ℃. The crystallization product was collected by terrorism to vacuum filtration. To obtain the desired compound after drying under reduced pressure at 45 ℃. White crystalline powder, 1.2g, 93% yield.
1 H-NMR (500 MHz, DMSO) delta (ppm) 1.35 (D, J = 8.83 Hz, 1H) 1.42-1.57 (M, 1H) 1.66-1.97 (M, 2H) 2.54-2.77 (M, 2H) 2.91 ( d, J = 11.35 Hz, 1H) 3.00-3.07 (m, 1H) 3.08 (m, 1H) 3.09 (s, 3H) 3.14 (m, 1H) 5.12 (d, J = 16.08 Hz, 1H) 5.20 (d, J = 16.39 Hz, 1H) 5.38 (s, 1H) 7.17 (dd, J = 9.62 Hz, 2.36 Hz, 1H) 7.35 (td, J = 8.51 Hz, 2.52 Hz, 1H) 7.95 (dd, J = 8.67 Hz, 5.52 Hz, 1H).
13 C NMR (126 MHz, DMSO) delta ppm 27.9, 31.6, 46.3, 47.0, 51.7, 55.8, 90.3, 106.9, 115.7, 117.1, 136.45, 136.53, 145.8, 152.3, 159.7, 162.7, 164.1 , 166.1, 175.2.

PATENT
PATENT
New Patent, Trelagliptin, SUN PHARMA
SUN PHARMACEUTICAL INDUSTRIES LIMITED [IN/IN]; Sun House, Plot No. 201 B/1 Western Express Highway Goregaon (E) Mumbai, Maharashtra 400 063 (IN)
BARMAN, Dhiren, Chandra; (IN).
NATH, Asok; (IN).
PRASAD, Mohan; (IN)
NATH, Asok; (IN).
PRASAD, Mohan; (IN)
The present invention provides a process for the preparation of 4-fluoro-2- methylbenzonitrile of Formula (II), and its use for the preparation of trelagliptin or its salts. The present invention provides an efficient, simple, and commercially friendly process for the preparation of 4-fluoro-2-methylbenzonitrile, which is used as an intermediate for the preparation of trelagliptin or its salts. The present invention avoids the use of toxic and hazardous reagents, high boiling solvents, and bromo intermediates such as 2-bromo-5-fluorotoluene, which is lachrymatory in nature and thus difficult to handle at a commercial scale.
Trelagliptin is a dipeptidyl peptidase IV (DPP-IV) inhibitor, chemically designated as 2- [[6-[(3i?)-3 -aminopiperidin- 1 -yl] -3 -methyl -2,4-dioxopyrimidin- 1 -yljmethyl] -4-fluorobenzonitrile, represented by Formula I.

Formula I
Trelagliptin is administered as a succinate salt of Formula la, chemically designated as 2-[[6-[(3i?)-3-aminopiperidin-l-yl]-3-methyl-2,4-dioxopyrimidin-l-yl]methyl]-4-fluorobenzonitrile butanedioic acid (1 : 1).

Formula la
U.S. Patent Nos. 7,795,428, 8,288,539, and 8,222,411 provide a process for the preparation of 4-fluoro-2-methylbenzonitrile by reacting 2-bromo-5-fluorotoluene with copper (I) cyanide in N,N-dimethylformamide.
Chinese Patent No. CN 102964196 provides a process for the preparation of 4-fluoro-2-methylbenzonitrile by reacting 4-fluoro-2-methylbenzyl alcohol with cuprous iodide in the presence of 2,2′-bipyridine and 2,2,6,6-tetramethylpiperidine oxide (TEMPO) in an anhydrous ethanol.
Copper (I) cyanide is toxic to humans, and therefore its use in the manufacture of a drug substance is not advisable. In addition, 2-bromo-5-fluorotoluene is converted to 4-fluoro-2-methylbenzonitrile by refluxing in N,N-dimethylformamide at 152°C to 155°C for 24 hours. This leads to some charring, resulting in a tedious work-up process and low yield. Furthermore, the use of reagents like cuprous iodide, 2,2′-bipyridine, and 2,2,6,6-tetramethylpiperidine oxide (TEMPO) is hazardous and/or environmentally-unfriendly, and therefore their use in the manufacture of a drug substance is not desirable.
The present invention provides an efficient, simple, and commercially friendly process for the preparation of 4-fluoro-2-methylbenzonitrile, which is used as an intermediate for the preparation of trelagliptin or its salts. The present invention avoids the use of toxic and hazardous reagents, high boiling solvents, and bromo intermediates such as 2-bromo-5-fluorotoluene, which is lachrymatory in nature and thus difficult to handle at a commercial scale.
EXAMPLES
Example 1 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (1.38 g) was added to ethanol (10 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (2.76 g) and pyridine (1 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 3 hours. The solvent was recovered up to maximum extent from the reaction mixture under reduced pressure to afford the title compound. Yield: 3.1 g
Example 2: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (5 g) was added to ethanol (37 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (10 g) and N,N-diisopropylethylamine (3.6 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 2 hours. The solvent was recovered up to maximum extent from the reaction mixture under reduced pressure to afford the title compound. Yield: 3.1 g
Example 3 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (10 g) was added to ethanol (40 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (20 g) and N,N-diisopropylethylamine (7.5 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to afford the title compound. Yield: 11.0 g
Example 4: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (50 g) was added to ethanol (500 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (70 g) and N,N-diisopropylethylamine (36 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 6 hours. The solvent was recovered from the reaction mixture under reduced pressure to afford the title compound. Yield: 51.0 g
Example 5 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (20 g) was added to ethanol (200 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (20 g) and N,N-diisopropylethylamine (18 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to obtain a residue. Deionized water (60 mL) was charged into the residue, and then the slurry was stirred at 0°C to 5°C for 1 hour. The solid obtained was filtered, then washed with deionized water (2 x 20 mL). The wet solid was dried in an air oven at 40°C to 45 °C for 4 hours to 5 hours. The crude product obtained was recrystallized in ethanol (50 mL) to afford the pure title compound. Yield: 21.0 g
Example 6: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methyl benzaldehyde (50 g) was added to ethanol (500 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (50 g) and N,N-diisopropylethylamine (46.4 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to obtain a residue. Deionized water (150 mL) was charged to the residue, and then the slurry was stirred at 0°C to 5°C for 1 hour. The solid obtained was filtered, then washed with deionized water (2 x 50 mL). The wet solid was dried in an air oven at 40°C to 45 °C for 4 hours to 5 hours. The crude product obtained was recrystallized in ethanol (200 mL) to afford the pure title compound. Yield: 53.5 g
Example 7: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (3.1 g) and phosphorous pentoxide (1 g) were added to toluene (30 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 24 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (30 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 1.1 g
Example 8: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (3 g) and phosphorous pentoxide (2 g) were added to toluene (30 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 24 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (30 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 1.0 g
Example 9: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (5 g) and concentrated sulphuric acid (2 mL) were added to toluene (100 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 5 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (50 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 3.24 g
Example 10: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (25 g) and concentrated sulphuric acid (35 g) were added to toluene (500 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 6 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (250 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 20.5 g
Example 11 : Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methyl benzaldoxime (5 g) and sodium bisulphate monohydrate (3.1 g) were added to toluene (50 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C, then filtered, and then washed with toluene (10 mL). The filtrate was concentrated under reduced pressure to afford the title compound. Yield: 3.0 g
Example 12: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methyl benzaldoxime (50 g) and sodium bisulphate monohydrate (31.6 g) were added to toluene (500 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C using a Dean-Stark apparatus for 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25 °C to 30°C, then filtered, and then washed with toluene (100 mL). The filtrate was concentrated under reduced pressure to afford a crude product. The crude product obtained was recrystallized in a mixture of toluene (200 mL) and hexane (500 mL) to afford the title compound.
Yield: 38.0 g


Sun Pharma managing director Dilip Shanghvi.
References
- 1 McKeage, Kate (2015-06-27). "Trelagliptin: First Global Approval". Drugs 75 (10): 1161–1164. doi:10.1007/s40265-015-0431-9. ISSN 0012-6667.
- 2 Inagaki, Nobuya; Onouchi, Hitoshi; Sano, Hiroki; Funao, Nobuo; Kuroda, Shingo; Kaku, Kohei. "SYR-472, a novel once-weekly dipeptidyl peptidase-4 (DPP-4) inhibitor, in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial". The Lancet Diabetes & Endocrinology 2 (2): 125–132. doi:10.1016/s2213-8587(13)70149-9.
- 3"New Drug Application Approval of Zafatek® Tablets for the treatment of Type 2 Diabetes in Japan | Takeda Pharmaceutical Company Limited". www.takeda.com. Retrieved 2015-11-13.
- 4 Cahn, Avivit; Raz, Itamar (2013-06-01). "Emerging gliptins for type 2 diabetes". Expert Opinion on Emerging Drugs 18 (2): 245–258. doi:10.1517/14728214.2013.807796. ISSN 1472-8214. PMID 23725569.
- 5Inagaki, Nobuya; Onouchi, Hitoshi; Maezawa, Hideaki; Kuroda, Shingo; Kaku, Kohei. "Once-weekly trelagliptin versus daily alogliptin in Japanese patients with type 2 diabetes: a randomised, double-blind, phase 3, non-inferiority study". The Lancet Diabetes & Endocrinology 3 (3): 191–197. doi:10.1016/s2213-8587(14)70251-7.
- 6"TRELAGLIPTIN SUCCINATE | C22H26FN5O6 - PubChem". pubchem.ncbi.nlm.nih.gov. Retrieved 2015-11-13.
- 7"Trelagliptin succinate | C22H26FN5O6 | ChemSpider". www.chemspider.com. Retrieved 2015-11-13.
| Patent | Submitted | Granted |
|---|---|---|
| TABLET [US2012129878] | 2010-07-27 | 2012-05-24 |
| AROMATIC RING COMPOUND [US2015045378] | 2013-02-12 | 2015-02-12 |
| Patent | Submitted | Granted |
|---|---|---|
| Combination therapy for the treatment of diabetes and related conditions [US2011263617] | 2011-10-27 | |
| Treatment for diabetes in patients with insufficient glycemic control despite therapy with an oral or non-oral antidiabetic drug [US2011275561] | 2011-11-10 | |
| Treatment for diabetes in patients with inadequate glycemic control despite metformin therapy comprising a DPP-IV inhibitor [US2011301182] | 2011-12-08 | |
| COATED PREPARATION [US2010166853] | 2008-07-10 | 2010-07-01 |
| Solid preparation comprising 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-4-fluorobenzonitrile [US7994183] | 2008-03-12 | 2011-08-09 |
| Diabetes therapy [US2012165251] | 2011-06-23 | 2012-06-28 |
| MEDICAL USE OF A DPP-4 INHIBITOR [US2014371243] | 2014-06-13 | 2014-12-18 |
| TREATMENT OF GENOTYPED DIABETIC PATIENTS WITH DPP-IV INHIBITORS SUCH AS LINAGLIPTIN [US2013196898] | 2010-11-26 | 2013-08-01 |
| ANTIDIABETIC MEDICATIONS COMPRISING A DPP-4 INHIBITOR (LINAGLIPTIN) OPTIONALLY IN COMBINATION WITH OTHER ANTIDIABETICS [US2012094894] | 2010-02-12 | 2012-04-19 |
| DPP-IV INHIBITORS FOR TREATMENT OF DIABETES IN PEDIATRIC PATIENTS [US2012122776] | 2010-01-29 | 2012-05-17 |
| Patent | Submitted | Granted |
|---|---|---|
| LAMINATED TABLET AND MANUFACTURING METHOD THEREFOR [US2014023708] | 2012-03-02 | 2014-01-23 |
| Combination therapy for the treatment of diabetes and related conditions [US2013310398] | 2013-07-24 | 2013-11-21 |
| USE OF KERATINOCYTES AS A BIOLOGICALLY ACTIVE SUBSTANCE IN THE TREATMENT OF WOUNDS, SUCH AS DIABETIC WOUNDS, OPTIONALLY IN COMBINATION WITH A DPP-4 INHIBITOR [US2013315975] | 2013-05-23 | 2013-11-28 |
| USE OF A DPP-4 INHIBITOR IN AUTOIMMUNE DIABETES, PARTICULARLY LADA [US2013317046] | 2013-05-21 | 2013-11-28 |
| USE OF A DPP-4 INHIBITOR FOR MODIFYING FOOD INTAKE AND REGULATING FOOD PREFERENCE [US2013324463] | 2013-05-21 | 2013-12-05 |
| COMBINATION THERAPY [US2013281373] | 2011-05-05 | 2013-10-24 |
| USE OF A DPP-4 INHIBITOR IN PODOCYTES RELATED DISORDERS AND/OR NEPHROTIC SYNDROME [US2013303462] | 2013-05-13 | 2013-11-14 |
| USE OF A DPP-4 INHIBITOR IN SIRS AND/OR SEPSIS [US2013303554] | 2013-05-13 | 2013-11-14 |
| Combination of a GPR119 Agonist and the DPP-IV Inhibitor Linagliptin for Use in the Treatment of Diabetes and Related Conditions [US2013109703] | 2011-03-18 | 2013-05-02 |
| Treatment for diabetes in patients inappropriate for metformin therapy [US2011263493] | 2011-10-27 |
| Patent | Submitted | Granted |
|---|---|---|
| DIPEPTIDYL PEPTIDASE INHIBITORS [US7781584] | 2008-07-03 | 2010-08-24 |
| POLYMORPHS OF SUCCINATE SALT OF 2-[6-(3-AMINO-PIPERIDIN-1-YL)-3-METHYL-2,4-DIOXO-3,4-DIHYDRO-2H-PYRIMIDIN-1-YLMETHY]-4-FLUOR-BENZONITRILE AND METHODS OF USE THEREFOR [US2008227798] | 2008-09-18 | |
| GPR119 receptor agonists in methods of increasing bone mass and of treating osteoporosis and other conditions characterized by low bone mass, and combination therapy relating thereto [US7816364] | 2009-10-29 | 2010-10-19 |
| DIPEPTIDYL PEPTIDASE INHIBITORS [US8222411] | 2009-11-05 | 2012-07-17 |
| ADMINISTRATION OF DIPEPTIDYL PEPTIDASE INHIBITORS [US2008287476] | 2008-11-20 | |
| POLYMORPHS OF SUCCINATE SALT OF 2-[6-(3-AMINO-PIPERIDIN-1-YL)-3-METHYL-2,4-DIOXO-3,4-DIHYDRO-2H-PYRIMIDIN-1-YLMETHY]-4-FLUOR-BENZONITRILE AND METHODS OF USE THEREFOR [US8084605] | 2008-11-13 | 2011-12-27 |
| WEEKLY ADMINISTRATION OF DIPEPTIDYL PEPTIDASE INHIBITORS [US8093236] | 2008-11-06 | 2012-01-10 |
| Therapeutic Agent for Diabetes [US2009042863] | 2009-02-12 | |
| ADMINISTRATION OF DIPEPTIDYL PEPTIDASE INHIBITORS [US2007060530] | 2007-03-15 | |
| DIPEPTIDYL PEPTIDASE INHIBITORS [US7795428] | 2008-01-03 | 2010-09-14 |
| Patent | Submitted | Granted |
|---|---|---|
| Dipeptidyl peptidase inhibitors [US7807689] | 2005-11-24 | 2010-10-05 |
| DIPEPTIDYL PEPTIDASE INHIBITORS [US2008108807] | 2008-05-08 | |
| DIPEPTIDYL PEPTIDASE INHIBITORS [US2008108808] | 2008-05-08 | |
| FUSED CYCLIC COMPOUNDS [US7732626] | 2010-01-07 | 2010-06-08 |
| DIPEPTIDYL PEPTIDASE INHIBITORS [US7906523] | 2008-08-07 | 2011-03-15 |
| DIPEPTIDYL PEPTIDASE INHIBITORS [US8188275] | 2008-07-24 | 2012-05-29 |
| DIPEPTIDYL PEPTIDASE INHIBITORS [US8173663] | 2009-01-08 | 2012-05-08 |
| ADMINISTRATION OF DIPEPTIDYL PEPTIDASE INHIBITORS [US2011077402] | 2011-03-31 | |
| DPP-IV INHIBITORS FOR USE IN THE TREATMENT OF NAFLD [US2011092510] | 2011-04-21 | |
| PURIN DERIVATIVES FOR USE IN THE TREATMENT OF FAB-RELATED DISEASES [US2011190322] | 2011-08-04 |
| Patent | Submitted | Granted |
|---|---|---|
| Administration of Dipeptidyl Peptidase Inhibitors [US2011192748] | 2011-08-11 | |
| PHARMACEUTICAL COMPOSITION COMPRISING A GLUCOPYRANOSYL-SUBSTITUTED BENZENE DERIVATE [US2011195917] | 2011-08-11 | |
| DPP-IV INHIBITOR COMBINED WITH A FURTHER ANTIDIABETIC AGENT, TABLETS COMPRISING SUCH FORMULATIONS, THEIR USE AND PROCESS FOR THEIR PREPARATION [US2011206766] | 2011-08-25 | |
| COMBINATION OF A CERTAIN DPP-4 INHIBITOR AND VOGLIBOSE [US2014343014] | 2014-05-16 | 2014-11-20 |
| CARDIO- AND RENOPROTECTIVE ANTIDIABETIC THERAPY [US2014274889] | 2014-03-14 | 2014-09-18 |
| TREATMENT FOR DIABETES IN PATIENTS INAPPROPRIATE FOR METFORMIN THERAPY [US2014274890] | 2014-06-03 | 2014-09-18 |
| Fused ring compound and use thereof [US2010190747] | 2010-07-29 | |
| FUSED RING COMPOUND AND USE THEREOF [US2010197683] | 2010-08-05 | |
| Fused cyclic compounds [US8088821] | 2010-08-05 | 2012-01-03 |
| GPR119 Receptor Agonists in Methods of Increasing Bone Mass and of Treating Osteoporosis and Other Conditions Characterized by Low Bone Mass, and Combination Therapy Relating Thereto [US8101626] | 2010-07-29 | 2012-01-24 |
 | |
| Systematic (IUPAC) name | |
|---|---|
| Succinic acid - 2-({6-[(3R)-3-amino-1-piperidinyl]-3-methyl-2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl}methyl)-4-fluorobenzonitrile (1:1) | |
| Clinical data | |
| Trade names | Zafatek |
| Chemical data | |
| Formula | C22H26FN5O6 |
| Molar mass | 475.470143 g/mol |
/////////Trelagliptin, PMDA, JAPAN 2015
Cn1c(=O)cc(n(c1=O)Cc2cc(ccc2C#N)F)N3CCC[C@H](C3)N
CN1C(=O)C=C(N(C1=O)CC2=C(C=CC(=C2)F)C#N)N3CCCC(C3)N