 .
.Picture credit....Bethany Halford
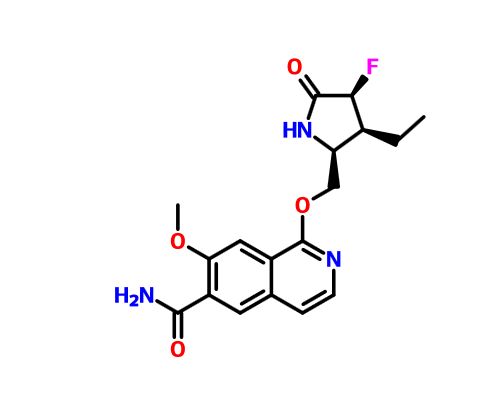
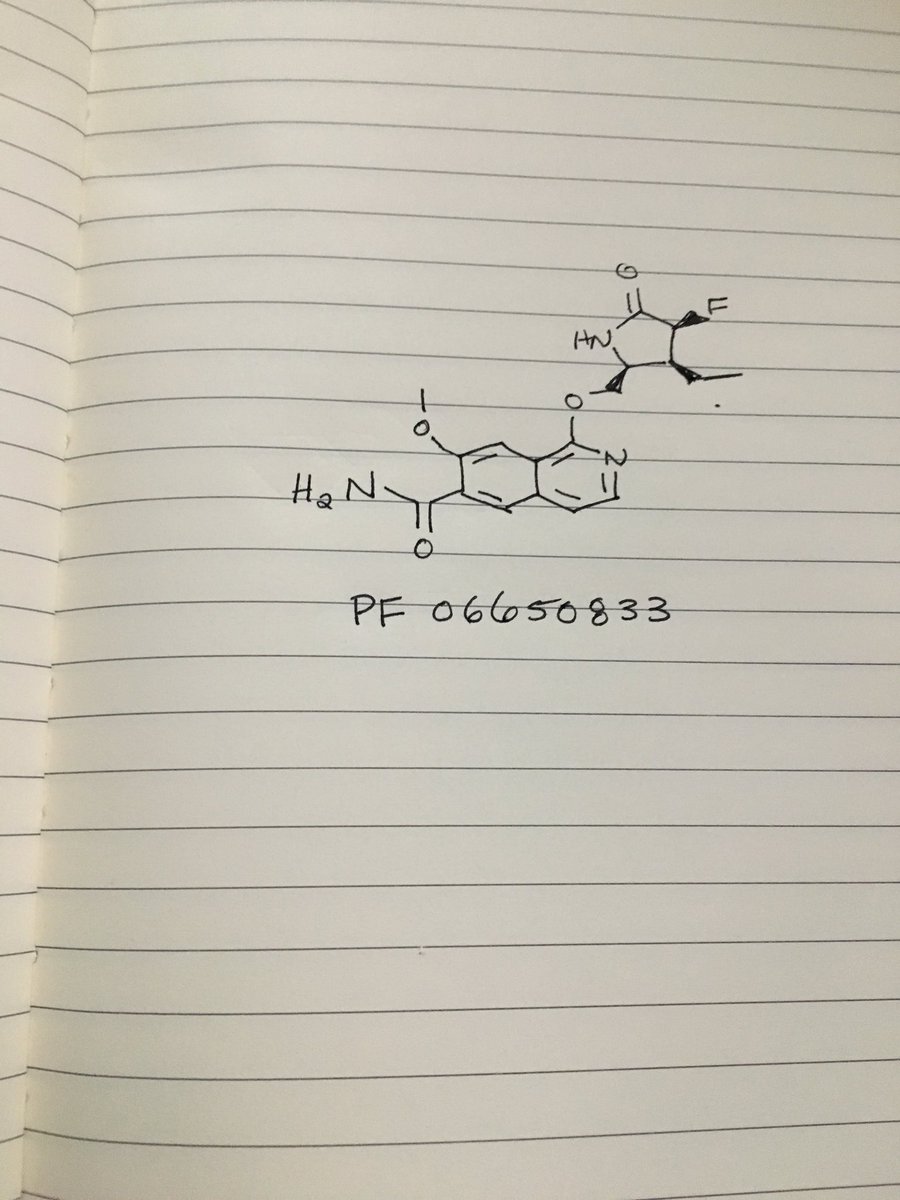
PF 06650833
MFC18H20FN3O4, MW361.371-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide
6-Isoquinolinecarboxamide, 1-[[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxo-2-pyrrolidinyl]methoxy]-7-methoxy-
CAS 1817626-54-2
WO 2015150995
1st disclosures is @pfizer's on inflammatory disease treatment targeting IRAK4
Phase I Lupus vulgaris
- 01 Feb 2016 Pfizer completes a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Nov 2015 Pfizer initiates a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Jun 2015 Pfizer completes a phase I trial for Lupus (In volunteers) in USA (PO) (NCT02224651)
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified in tyrosine and serine/threonine kinases. Inappropriate activity arising from dysregulation of certain kinases by a variety of mechanisms is believed to underlie the causes of many diseases, including but not limited to, cancer, cardiovascular diseases, allergies, asthma, respiratory diseases, autoimmune diseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative diseases. As such, potent and selective inhibitors of kinases are sought as potential treatments for a variety of human diseases.
There is considerable interest in targeting the innate immune system in the treatment of autoimmune diseases and sterile inflammation. Receptors of the innate immune system provide the first line of defense against bacterial and viral insults. These receptors recognize bacterial and viral products as well as pro-inflammatory cytokines and thereby initiate a signaling cascade that ultimately results in the up-regulation of inflammatory cytokines such as TNFa, IL6, and interferons. Recently it has become apparent that self-generated ligands such as nucleic acids and products of inflammation such as high-mobility group protein B1 (HMGB1) and Advanced Glycated End-products (AGE) are ligands for Toll-like receptors (TLRs) which are key receptors of the innate immune system (O'Neill 2003, Kanzler et al 2007, Wagner 2006). This demonstrates the role of TLRs in the initiation and perpetuation of inflammation due to autoimmunity.
lnterleukin-1 receptor associated kinase 4 (I RAK4) is a ubiquitously expressed serine/threonine kinase involved in the regulation of innate immunity (Suzuki & Saito 2006). IRAK4 is responsible for initiating signaling from TLRs and members of the I L- 1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice were reported to cause reductions in TLR and IL-1 induced pro-inflammatory cytokines (Kawagoe et al 2007; Fraczek et al. 2008; Kim et al. 2007). IRAK4 kinase-dead knock-in mice have also been shown to be resistant to induced joint inflammation in the antigen-induced-arthritis (AIA) and serum transfer-induced (K/BxN) arthritis models (Koziczak-Holbro 2009). Likewise, humans deficient in IRAK4 also appear to display the inability to respond to challenge by Toll ligands and IL-1 (Hernandez & Bastian 2006). However, the immunodeficient phenotype of IRAK4-null individuals is narrowly restricted to challenge by gram positive bacteria, but not gram negative bacteria, viruses or fungi. This gram positive sensitivity also lessens with age, implying redundant or compensating mechanisms for innate immunity in the absence of IRAK4 (Lavine et al 2007).
These data indicate that inhibitors of IRAK4 kinase activity should have therapeutic value in treating cytokine driven autoimmune diseases while having minimal immunosuppressive side effects. Additional recent studies suggest that targeting IRAK4 may be useful in other inflammatory pathologies such as atherosclerosis and diffuse large B-cell lymphoma (Rekhter et al 2008; Ngo et al 2011). Therefore, inhibitors of IRAK4 kinase activity are potential therapeutics for a wide variety of diseases including but not limited to autoimmunity, inflammation, cardiovascular diseases, cancer, and metabolic diseases. See the following references for additional information: N. Suzuki and T. Saito, Trends in Immunology, 2006, 27, 566. T. Kawagoe, S. Sato, A. Jung, M. Yamamoto, K. Matsui, H. Kato, S. Uematsu, O. Takeuchi and S. Akira, Journal of Experimental Medicine, 2007, 204, 1013. J. Fraczek, T. W. Kim, H. Xiao, J. Yao, Q. Wen, Y. Li, J.-L. Casanova, J. Pryjma and X. Li, Journal of Biological Chemistry, 2008, 283, 31697. T. W. Kim, K. Staschke, K. Bulek, J. Yao, K. Peters, K.-H. Oh, Y. Vandenburg, H. Xiao, W. Qian, T. Hamilton, B. Min, G. Sen, R. Gilmour and X. Li, Journal of Experimental Medicine, 2007, 204, 1025. M. Koziczak-Holbro, A. Littlewood- Evans,
B. Pollinger, J. Kovarik, J. Dawson, G. Zenke, C. Burkhart, M. Muller and H. Gram, Arthritis & Rheumatism, 2009, 60, 1661. M. Hernandez and J. F. Bastian, Current Allergy and Asthma Reports, 2006, 6, 468. E. Lavine, R. Somech, J. Y. Zhang, A. Puel, X. Bossuyt, C. Picard, J. L. Casanova and C. M. Roifman, Journal of Allergy and Clinical Immunology, 2007, 120, 948. M. Rekhter, K. Staschke, T. Estridge, P. Rutherford, N. Jackson, D. Gifford-Moore, P. Foxworthy,
C. Reidy, X.-d. Huang, M. Kalbfleisch, K. Hui, M.S. Kuo, R. Gilmour and C. J. Vlahos, Biochemical and Biophysical Research Communications, 2008, 367, 642. O'Neill, L. A. (2003). "Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases." Curr Opin Pharmacol 3(4): 396. Kanzler, H et al. (2007) "Therapeutic targeting of innate immunity with toll-like receptor agonists and antagonists." Nature Medicine 13:552. Wagner, H. (2006) "Endogenous TLR ligands and autoimmunity" /Advances in Immunol 91 : 159. Ngo, V. N. et al. (2011) "Oncogenically active MyD88 mutations in human lymphoma" Nature 470: 115.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015150995&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
Preparation 1 : 1-chloro-7-methoxyisoquinoline-6-carbonitrile (P1) Step 1. Synthesis of methyl 4-iodo-3-methoxybenzoate (CAS 35387-92-9. CD.
To a solution of 3-hydroxy-4-iodobenzoic acid (CAS 58123-77-6, C12) (10800 g, 40.9 moles) in DMF (65 L) was added K2C03 (25398 g, 184 moles), followed by the slow addition of dimethyl sulfate (11352 g, 90 moles). This mixture was heated to about 50 °C for over night. The reaction mixture was cooled to about 25 °C, diluted with EtOAc (50 L) and filtered through a plug of Celite®. The solid was thoroughly washed with EtOAc (10 L X 3). The combined EtOAc filtrates were poured into water. After stirring for about 30 min, the EtOAc layer was separated and it was further washed sequentially with water, 1 M NaOH and brine. The EtOAc layer was separated, dried over Na2S04, filtered and concentrated to provide the title compound C1. Yield: 11750 g (98%).
Step 2. Synthesis of (4-iodo-3-methoxyphenyl)methanol (CAS 244257-61-2, C2).
To a solution of compound C1 (11750 g, 40.2 moles) in THF (35 L) was added NaBH4 (7645 g, 201.09 moles) and refluxed. While refluxing, MeOH (25 L) was slowly added into the reaction mixture at a rate of about 1 L per hour. After completion of the reaction, it was poured into a solution of cold dilute HCI. Once the excess of NaBH4was quenched, the solution was filtered and extracted with EtOAc (2.5 L X 3). The combined EtOAc extracts were washed sequentially with water, brine and dried over Na2S04. The solvent was evaporated under reduced pressure and the resulting crude material was treated with MTBE. The resulting solid was filtered and filtrate was washed with water, brine, dried over Na2S0 , and filtered. The solvent was evaporated under reduced pressure to provide the title compound C2. Yield: 9900 g (93%).
Step 3. Synthesis of 4-iodo-3-methoxybenzaldehyde (CAS 121404-83-9, C3).
To a solution of compound C2 (9900 g, 34.5 moles) in CHCI3 (186 L), was added manganese dioxide (18000 g, 207 moles) and the resulting mixture was refluxed for about 16 h. The mixture was cooled to about 25 °C and filtered through a Celite pad, which was then washed thoroughly with CHCI3. The CHCI3 was evaporated under reduced pressure to provide the title compound C3. Yield: 9330 g (95%). 1 H NMR (400 MHz, CDCI3): δ 9.95 (s, 1 H), 7.99 (d, 1 H), 7.14 (dd, 1 H), 3.95 (s, 3 H).
Step 3. Synthesis of 6-iodo-7-methoxyisoquinoline (CAS 244257-63-4. C4).
To a solution of compound C3 (9300 g, 35 moles) in toluene (60 L) was added amino acetaldehyde dimethyl acetal (5590 g, 53 moles) and the mixture was refluxed for about 4 h, while removing the liberated water by the use of a Dean - Stark water separator. The reaction mixture was cooled to about 0 °C, after which trifluoroacetic anhydride (22305 g, 106 moles) followed by BF3-Et20 (15080 g, 106 moles) were added, keeping internal temperature below 5 °C. The reaction mixture was stirred at about 25 °C for about 16 h and quenched by pouring into a mixture of ice and ammonium hydroxide. The product was extracted with EtOAc (10 L X 3), and the combined EtOAc extracts were washed sequentially with water and brine. The combined EtOAc extracts were dried over Na2S04, filtered, and concentrated to afford a dark tan colored residue. This was treated with a mixture of MTBE and hexane (1 :1 v/v, 30 L), followed by 6 M HCI (9 L), with stirring. The precipitated solid was filtered and washed with MTBE. The solid was suspended in EtOAc (5 L) and made alkaline with ammonium hydroxide. The EtOAc layer was separated, washed with brine, dried over Na2S04, filtered, and concentrated to afford crude compound C4 as a brown solid. HPLC (230 nm) showed it to be about 83% pure.
The crude material (1000 g) was taken in AcOH (2.5 L) and stirred for about 90 min at about 25 °C. The solid was filtered and washed with AcOH (500 ml_). The filtrate was neutralized with saturated aqueous Na2C03 solution. The resulting precipitated solid was filtered, washed with water (4 L), and oven dried at about 70 - 75 °C for about 5 h to afford about 780 g of pure C4. Similarly, the remaining crude C4 (4 kg) was purified to provide the title compound C4. Yield: 4300 g (42%). 1H NMR (400 MHz, CDCI3): δ 9.15 (s, 1 H), 8.45 (d, 1 H), 8.35 (s, 1 H), 7.45 (d, 1 H), 7.15 (s, 1 H) 4.00 (s, 3 H).
Step 4. Synthesis of 7-methoxyisoquinoline-6-carbonitrile (C5).
To a solution of compound C4 (4300 g , 15 moles) in DMSO (39 L) was added copper(l) cyanide (2954 g, 33 moles) and the mixture was heated to about 120 °C for about 3 h. The reaction mixture was quenched by pouring into a mixture of ice and ammonium hydroxide (40 L) and filtered. The filtrate was extracted with EtOAc (10 L X 2). While stirring, the solid residue was again treated with ammonium hydroxide solution (10 L) and EtOAc (10 L). After filtration, the precipitated material was repeatedly washed with a mixture of MeOH and CHCI3 (1 :9, v/v) several times and the combined extracts were washed with brine. The extracts were dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting crude material was triturated with hexane to provide the title compound C5. Yield: 2250 g (87%). 1H NMR (400 MHz, CDCI3): δ 9.25 (br. s, 1 H), 8.55 (br. s, 1 H), 8.15 (s, 1 H), 7.60 (d, 1 H), 7.30 (s, 1 H), 4.05 (s, 3 H).

A solution of a reactant such as 1-(((2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl)methoxy)-7-methoxyisoquinoline-6-carbonitrile (200 mg, 0.5 mmol) in concentrated H2SO4 (1.5 ml.) was warmed to about 55 °C for about two hours, then cooled to about 20 °C. The reaction mixture was added dropwise with vigorous stirring to 7.3 ml_ of ice cold concentrated ammonium hydroxide with cooling in ice. The precipitated solid was filtered and washed with water, heptane, ether, and dried under vacuum. The residue may be used directly for subsequent work, or it may be purified by chromatography or HPLC.
ABSTRACTS
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 261




//////////PF 06650833, IRAK4 inhibitor, inflammatory disease treatment , PFIZER, 1817626-54-2
N1C([C@H](C([C@H]1COc3c2cc(c(cc2ccn3)C(=O)N)OC)CC)F)=O
NC(=O)c2cc3ccnc(OC[C@H]1NC(=O)[C@@H](F)[C@H]1CC)c3cc2OC
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
P.STHE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
 .
.




















