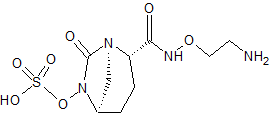

RG-6080
Sulfuric acid, mono[(1R,2S,5R)-2-[[(2-aminoethoxy)amino]carbonyl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
Phase I
A β-lactamase inhibitor potentially for the treatment of bacterial infections.
RG-6080; FPI-1459; OP-0595
CAS No. 1452458-86-4
| Molecular Formula | C9 H16 N4 O7 S |
| Formula Weight | 324.31 |
- Originator Fedora Pharmaceuticals
- Developer Meiji Seika Pharma
- Class Antibacterials; Azabicyclo compounds
- Mechanism of Action Beta lactamase inhibitors
- Phase IBacterial infections
Most Recent Events
- 13 Jan 2015 OP 0595 licensed to Roche worldwide, except Japan ,
- 30 Nov 2014 Meiji Seika Pharma completes a phase I trial in Healthy volunteers in Australia (NCT02134834)
- 01 May 2014 Phase-I clinical trials in Bacterial infections (in volunteers) in Australia (IV)

SYNTHESIS
WO 2015046207,
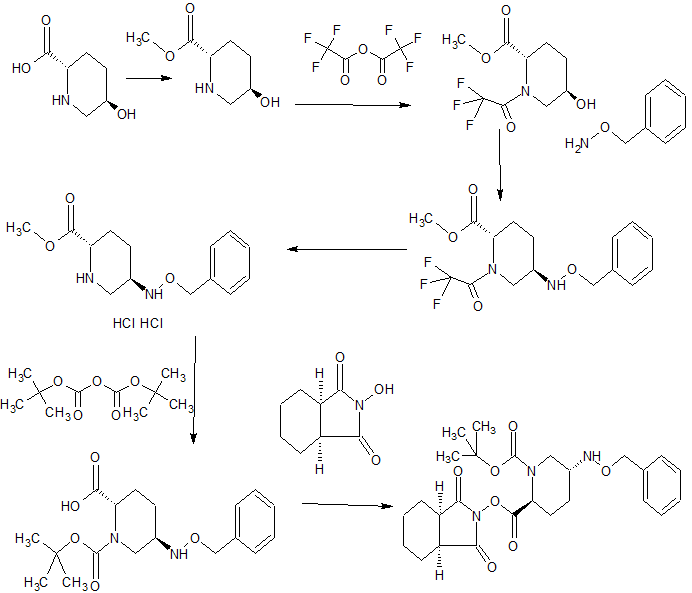
CONTD.......................
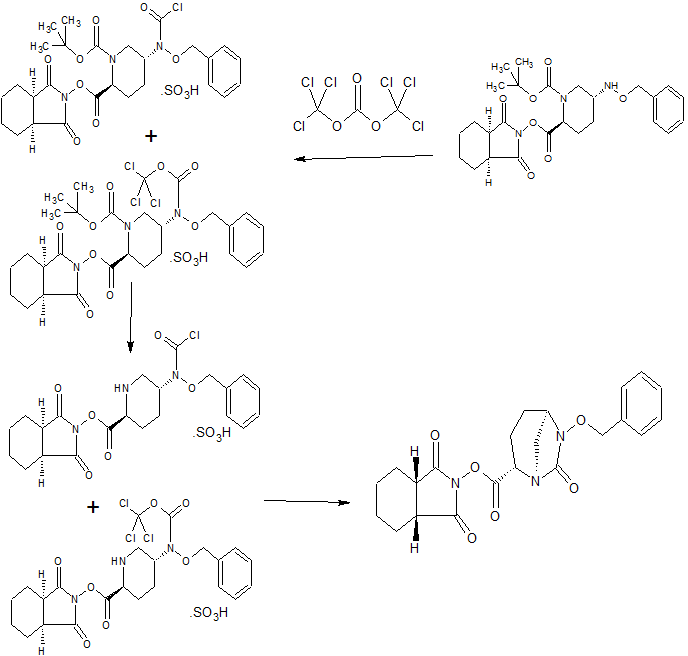
CONTD......................................
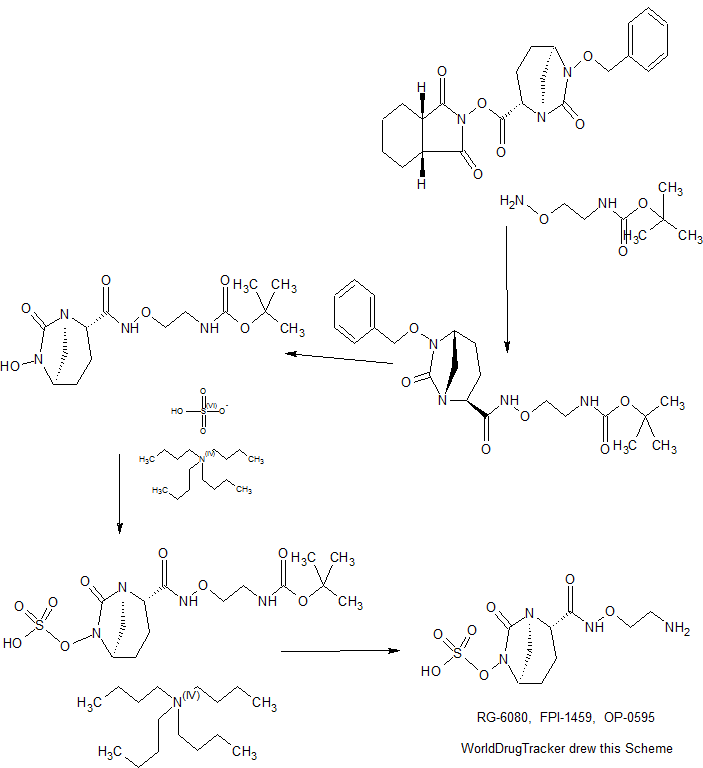
Patent
The novel heterocyclic compound in Japanese Patent 4515704 (Patent Document 1), preparation and shown for their pharmaceutical use, sodium trans-7-oxo-6- (sulfooxy) as a representative compound 1,6-diazabicyclo [3 .2.1] discloses an octane-2-carboxamide (NXL104). Preparation in regard to certain piperidine derivatives which are intermediates Patent 2010-138206 (Patent Document 2) and JP-T 2010-539147 (Patent Document 3) are shown at further WO2011 / 042560 (Patent Document 4) NXL104 to disclose a method for producing the crystals.
In Patent 5038509 (Patent Document 5) (2S, 5R) -7- oxo -N- (piperidin-4-yl) -6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane - 2- carboxamide (MK7655) is shown, discloses the preparation of certain piperidine derivatives with MK7655 at Patent 2011-207900 (Patent Document 6) and WO2010 / 126820 (Patent Document 7).
The present inventors also disclose the novel diazabicyclooctane derivative represented by the following formula (VII) in Japanese Patent Application 2012-122603 (Patent Document 8).
Patent Document 1: Japanese Patent No. 4515704 Pat
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
[Chemical formula 1] (In the formula, R 3 are the same as those described below)

Reference Example
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
step 1 tert-butyl {2 - [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate (IV-1)(2S, 5R)-6-(benzyloxy) -7-oxo-1,6-diazabicyclo [3.2.1] octane-2-carboxylic acid (4 .30g, dehydrated ethyl acetate (47mL) solution of 15.56mmol) was cooled to -30 ℃, isobutyl chloroformate (2.17g, washing included dehydration ethyl acetate 1mL), triethylamine (1.61g, washing included dehydration ethyl acetate 1 mL), successively added dropwise, and the mixture was stirred 1 hour at -30 ° C.. To the reaction solution tert- butyl 2-dehydration of ethyl acetate (amino-oxy) ethyl carbamate (3.21g) (4mL) was added (washing included dehydration ethyl acetate 1mL), raising the temperature over a period of 1.5 hours to 0 ℃, It was further stirred overnight. The mixture of 8% aqueous citric acid (56 mL), saturated aqueous sodium bicarbonate solution (40 mL), sequentially washed with saturated brine (40 mL), dried over anhydrous magnesium sulfate, filtered, concentrated to 5 mL, up to 6mL further with ethanol (10 mL) It was replaced concentrated. Ethanol to the resulting solution (3mL), hexane the (8mL) in addition to ice-cooling, and the mixture was stirred inoculated for 15 minutes. The mixture was stirred overnight dropwise over 2 hours hexane (75 mL) to. Collected by filtration the precipitated crystals, washing with hexane to give the title compound 5.49g and dried in vacuo (net 4.98 g, 74% yield). HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 50/50, 1.0 mL / min, UV 210 nm, Retweeted 4.4 min; 1 H NMR (400 MHz, CDCl 3 ) [delta] 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m, 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br.d., J = 11.6 Hz , 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br. . s, 1H), 7.34-7.48 (yd, 5H), 9.37 (Br.S., 1H); MS yd / z 435 [M + H] + .

Step 2
tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate
(V-1) tert-butyl {2 - [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
(V-1) tert-butyl {2 - [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
Step 3
Tetrabutylammonium tert- butyl {2 - [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide - pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
Tetrabutylammonium tert- butyl {2 - [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide - pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
Step 4 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
tetra butylammonium tert- butyl {2 - [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
tetra butylammonium tert- butyl {2 - [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
PATENT
Example
64 tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]

64 tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]
tert- butyl {2 - [({[(2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate (example 63q, net 156.42g, 360mmol) in methanol solution (2.4L) of 10% palladium carbon catalyst (50% water, 15.64g) was added, under an atmosphere of hydrogen, stirred for 1.5 hours did. The catalyst was filtered through celite, filtrate was concentrated under reduced pressure until 450mL, concentrated to 450mL by adding acetonitrile (1.5 L), the mixture was stirred ice-cooled for 30 minutes, collected by filtration the precipitated crystals, washing with acetonitrile, and vacuum dried to obtain 118.26g of the title compound (net 117.90g, 95% yield). Equipment data of the crystals were the same as those of the step 2 of Reference Example 3.
Example
65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)
[of 125]

65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)
[of 125]
tert- butyl {2 - [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (example 64,537.61g, 1.561mol) in acetonitrile (7.8L) solution of 2,6-lutidine (512.08g), sulfur trioxide - pyridine complex (810.3g) was added, at room temperature in the mixture was stirred overnight. Remove insolubles and the mixture was filtered, the filtrate concentrated to 2.5 L, diluted with ethyl acetate (15.1L). The mixture was extracted with 20% phosphoric acid 2 hydrogencarbonate aqueous solution (7.8L), the resulting aqueous layer into ethyl acetate (15.1L), added tetrabutylammonium hydrogen sulfate (567.87g), was stirred for 20 min. The organic layer was separated layers, dried over anhydrous magnesium sulfate (425 g), after filtration, concentration under reduced pressure, substituted concentrated tetrabutylammonium tert- butyl with dichloromethane (3.1L) {2 - [({[(2S, 5R ) -7-oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 758g (net 586.27g, Osamu rate 84%).
The tetra-butyl ammonium salt 719g (net 437.1g, 0.656mol) in dichloromethane (874mL) solution was cooled to -20 ℃, dropping trifluoroacetic acid (874mL) at 15 minutes, 1 the temperature was raised to 0 ℃ It was stirred time. The reaction was cooled to -20 ° C. was added dropwise diisopropyl ether (3.25L), and the mixture was stirred for 1 hour the temperature was raised to 0 ° C.. The precipitate is filtered, washed with diisopropyl ether to give the title compound 335.36g of crude and vacuum dried (net 222.35g, 99% yield).
The title compound of crude were obtained (212.99g, net 133.33g) and ice-cold 0.2M phosphate buffer solution of pH5.3 mix a little at a time, alternating between the (pH6.5,4.8L). The solution was concentrated under reduced pressure to 3.6L, it was adjusted to pH5.5 at again 0.2M phosphate buffer (pH6.5,910mL). The solution resin purification (Mitsubishi Kasei, SP207, water ~ 10% IPA solution) is subjected to, and concentrated to collect active fractions, after lyophilization, to give the title compound 128.3 g (96% yield). Equipment data of the crystals were the same as those of step 3 of Reference Example 3.
PATENT
US 20140288051
WO 2014091268
WO 2013180197
US 20130225554
///////////RG-6080, 1452458-86-4, FPI-1459, OP-0595, Phase I , β-lactamase inhibitor, bacterial infections, Fedora parmaceuticals, Meiji Seika Pharma