

ArgatrobanMolecular Formula: C23H36N6O5S
Formula Weight: 508.63
CAS No.: 74863-84-6
Formula Weight: 508.63
CAS No.: 74863-84-6
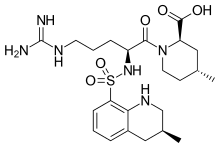
(2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2-
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl]
sulfonylamino]pentanoyl]-4-methyl-piperidine-2-
carboxylic acid
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl]
sulfonylamino]pentanoyl]-4-methyl-piperidine-2-
carboxylic acid
PATENT
US 7,589,106, 7,687,516, EP 0008746; US 4258192, US 4201863
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in the liver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).

Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.
……………………………………………………….
Argatroban monohydrate

Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5-[(aminoiminomethyl) amino]-1-oxo2[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4-methyl-2- piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. cas 141396-28-3
Argipidine, Argatroban monohydrate, GN1600, DK-7419, MDI-805 Acova, Slonnon, Novastan
Mitsubishi Chemical (Originator), Encysive Pharmaceuticals (Licensee), Mitsubishi Pharma (Distributor), Daiichi Pharmaceutical (Codevelopment), GlaxoSmithKline (Codevelopment), Mitsubishi Pharma (Codevelopment), Sanofi-SynthLabo (Codevelopment)
Antithrombocytopenic, CARDIOVASCULAR DRUGS, Cerebrovascular Diseases, Treatment of, HEMATOLOGIC DRUGS, Hematopoiesis Disorders Therapy, Ischemic Stroke, Treatment of, NEUROLOGIC DRUGS, Peripheral Vascular Disease, Treatment of, Stroke, Treatment of, Treatment of Peripheral Obstructive Vascular Disease, Thrombin Inhibitors
Synthesis of argatroban on the method reported in the literature there are two synthetic routes, patent EP8746, US4258192, US4201863, JP8115267 relates to a route is: with 4 – methyl-piperidine as a starting material was prepared first intermediate body (2R, 4R) -4 – methyl-2 – ethyl-piperidine, and the first and a t-BOC protected amino nitro-L-arginine condensation, and then the 3 – methyl – 8 – quinoline sulfonyl chloride condensation after hydrolysis, hydrogenation, hydration be argatroban. This entry route synthesis process complicated procedure to be carried out under the protection of nitrogen, the raw material is highly toxic gas phosgene, the operation more difficult.
US4117127, JP02-212473, EP823430, EP8746, JC S Perk Transl 1981 (5), JP02-212473 relates to an alternative route is: nitro L-arginine prior to the 3 – methyl-8 – subsequent condensation quinoline sulfonyl chloride Intermediate (2R, 4R) -4 – methyl-2 – piperidinecarboxylate condensation, and then after hydrolysis, hydrogenation, hydration be argatroban. This synthetic route despite the relatively simple process method, to obtain raw materials, but this method using reagents such as phosphorus oxychloride, phosphorus trichloride has a pungent odor, easy to absorb moisture in humid air, intense smoke, environmental pollution, greater stimulation of the body’s respiratory tract, can cause eye and skin irritation and burning, and the use of this method, complex operation, low yield, high cost.
Patent CN100586946C Argatroban discloses a method for separating optically active isomeric compounds, the feedstock argatroban mixed solvent of alcohol and water was heated to reflux 5-10 hours, cooled and allowed to stand, and filtered to give White crystalline product, dried, repeated 2-6 times. [0008] Patent CN101033223A discloses a Argatroban is the main by-product (2R, 4R)-l_ [N2-(3_ methyl-8 – quinolinesulfonyl)-L-arginyl] -4 – methyl-2 – carboxylic acid, argatroban, and the byproducts are difficult to isolate, argatroban two diastereomeric isomer 21 (S) and 21 (R) separation of work attracted a lot of research persons. Because both physical and chemical properties are very similar, so separation is very difficult. 1993 Rawson, Thomas E.; VanGorp, Kimmie A.; Yang, Janet so first by high pressure liquid chromatography and column chromatography separation to obtain a single 21 (S) and 21 (R) argatroban [ Journal of Pharmaceutical Sciences vol. 82, No. 6,672]; Thibaudeau Karen et al. reported Protein A chromatographic separation [US6440417]. However, due to the separation of these methods a small amount of low efficiency, so there is no practical value industrialization. 2006 China Tianjin Weijie Technology Co., Ltd. Song Honghai et al. Reported using recrystallization Separation 21 (S) and 21 (R) argatroban way [0 With 951,936 it], so that the mass 21 (5) Aga music classes as possible, but the law of low yield, complicated operation, high cost, and a large amount of a small amount of 21 (S) of 21 (R)-product argatroban, from the viewpoint of industrial production, is still a ideal method. [0009] These methods can be effectively prepared argatroban, but the purity of the desired product is not high, poor color, content is low, affecting the quality of the results of its preparation.
U.S. Pat. No. 4,201,863 (6 May 1980) and EP 8746 (filed on 22 Aug. 1979 with priority based on the application for the cited US patent) describe a class of N2-arylsulphonyl-L-argininamide drugs, with anti-thrombotic activity, and the processes for obtaining them. Of these, the compound 4-methyl-1-[N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid (argatroban, isomers mixture) is described. The described process comprises the synthesis of an intermediate NG-substituted-N2-quinolinesulphonyl-L-argininamide from which the desired compound is obtained by catalyzed hydrogenolysis or acidolysis and catalyzed hydrogenation. The general conditions provided for the hydrogenolysis and hydrogenation reaction are: i) inert solvents (methanol, ethanol, tetrahydrofuran or dioxane); ii) presence of a catalyst (Raney nickel, palladium, platinum, ruthenium, rhodium); iii) hydrogen atmosphere at a pressure between 1 and 100 kg/cm2 and preferably between 5 and 50 kg/cm2; iv) temperature between 0° C. and 200° C. and preferably between 50° C. and 150° C.; v) reaction temperature from 2 hours to 120 hours. The crude product obtained is then purified by trituration or by re-crystallization from diethyl ether-tetrahydrofuran, diethyl ether-methanol or from water-methanol or by chromatography. No example is given of this purification step. In particular, both U.S. Pat. No. 4,210,863 and EP 8746 in example 1(E) describe the preparation of argatroban, isomers mixture. This compound is obtained in amorphous form by hydrogenation of [NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid in ethanol in the presence of Pd/C with hydrogen pressure of 10 kg/cm2 at 100° C. for 8 hours. The catalyst is removed by filtration of the ethanol solution which is then evaporated without further purification and/or re-crystallization steps. In the US patent at issue as indeed in patent application EP 8746, no mention is made of polymorphic forms of the compounds and, for the obtained compound, the following characteristics are reported: Amorphous solid, I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.01; H 6.98; N 16.61.
U.S. Pat. No. 4,258,192 (24 Mar. 1981) (continuation-in-part of the aforesaid patent application U.S. Pat. No. 4,201,863) and the same patent application EP 8746 describe the stereoisomers and the preparation thereof, including argatroban used as an active principle in medicaments, i.e. the stereoisomer (2R,4R)-4methyl-[4N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid, with the following characteristics: melting point (m.p.). 188-191° C.; I.R. (KBr) (cm−1) 3400, 1620, 1460, 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.05; H 6.94; N 16.65. The compound is prepared according to the description given in examples 1(E) in U.S. Pat. No. 4,258,192 and 2(E) and 3 in EP 8746 respectively by hydrogenation of (2R,4R) 1-[NG-nitro-N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid in ethanol in presence of acetic acid catalyzed by Pd/C. After filtering the mass to remove the catalyst, the solvent is evaporated and the residue suspended in chloroform, the solution treated with a saturated sodium bicarbonate solution or 1N sodium hydroxide solution and after washing, the solvent is evaporated. The compound is then re-crystallized from ethanol. Again in this case, no reference is made to the obtainment of monohydrate polymorphic forms.
Said polymorphic forms are described instead in the publication Biochem. Biophys. Res. Comm. 1981, 101, 440-446 in the context of stereoisomer preparation. The monohydrate polymorph of the (2R,4R) stereoisomer is prepared by re-crystallization from ethanol/water and the reported characteristics are: m.p. 176-180° C.; [α]D 27 +76.1° (c 1, 0.2N HCl).
U.S. Pat. No. 5,925,760 (20 Jul. 1999) and EP 0823430 (filed 4 Aug. 1997) subsequently describe a new method for preparing argatroban by means of a new intermediate N2-(3-methyl-8-quinolinesulphonyl)-NG-nitro-L-arginine. In particular the patent makes reference to the preparation of a crystalline monohydrate form of argatroban, referring back to examples (D) and (E) of Japanese patent publication No. (Hei)-2-31055/1990 and generically to an I.R. spectrum identical to that of the commercially available argatroban compound. The relevant example in the cited patent publication is example (E), while example (D) concerns the preparation of (2R,4R)-1-[NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid. This compound represents the starting compound for argatroban preparation by catalytic reduction in the presence of Pd/C. The crude argatroban obtained is then purified by extraction with chloroform, treatment with a saturated sodium bicarbonate solution and, after solvent evaporation, re-crystallization from ethanol or from 15% alcohol in water. It should be noted however that the Japanese patent makes no mention of the monohydrate form of argatroban being obtained and that for the compound the following characteristics are reported: m.p. 188-191° C.; molecular composition (theoretical/found) (%): C 54.31/54.01; H 7.13/6.98; N 16.52/16.61; I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380. These analytical data, with the exception of the unreported melting point, are the same as those indicated in the cited patent documents describing a mixture of (2R,4R)-4methyl-[4N2-(3S-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid and (2R,4R)-4methyl-1-[N2-(3R-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid isomers of argatroban, but do not correspond to the melting point given in the publication, being the only document that identifies the monohydrate form of argatroban.
More recently, patent application CN 1,951,937 (filing date 10 Nov. 2006) described a method for preparing hydrated argatroban by treating argatroban with large quantities of water (more than 60 and up to 80 volumes of distilled water per gram of argatroban) at a temperature of 80-100° C. for a time of 0.5-1 hour and crystallization by cooling. The water content reported is comprised between 3.3 and 3.8% and the ratio of dextroisomer R to levoisomer S is R:S=63-67: 37-33.
Argatroban is a compound of wide therapeutic use, for which reason the need still exists to provide a compound of pharmaceutically acceptable quality obtained by easily industrialized and economically convenient methods. With regard to the monohydrate, this form is preferable for the applicative purpose since the anhydrous form is unstable and tends to become hydrated and/or wet. Moreover it crystallizes only with difficulty at the correct ratio between the diastereoisomers.

…..

the protection of 4-methylpiperidine (I) with (Boc)2O gives the carbamate (II), which is condensed with benzyl chloroformate by means of sec-butyl lithium and TMEDA in ethyl ether to yield (?-trans-1-(tert-butoxycarbonyl)-4-methylpiperidine-2-carboxylic acid benzyl ester (III). Deprotection of the NH group of (III) with HCl in ethyl acetate affords (?-trans-4-methylpiperidine-2-carboxylic acid benzyl ester (IV), which is condensed with the protected arginine derivative (V) by means of isobutyl chloroformate and TEA to provide the corresponding amide as a diastereomeric mixture. Resolution of this mixture by flash chromatography furnishes the desired diastereomer (VI), which is treated with HCl in ethyl acetate in order to remove the Boc-protecting group to yield compound (VII). Condensation of compound (VII) with 3-methylquinoline-8-sulfonyl chloride (VIII) by means of TEA in dichloromethane affords the expected sulfonamide (IX). Finally, this compound is submitted to hydrogenation with H2 over Pd/C in AcOH/ethanol in order to produce debenzylation, cleavage of the NO2 group and hydrogenation of the pyridine ring to yield argatroban.
………….
Argatroban, i.e., (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino) pentyl)-4-methyl-2-piperidine carboxylic acid, has two diastereoisomers: 21(R) and 21(S). Usually the ratio of 21(R) to 21(S) is 64-65: 36-35 (U.S. Pat. No. 6,440,417, Cossy. J., et al, Bioorganic & Medicine Chemistry Letters, 11 (2001), 1989-1992, Journal of pharmaceutical Sciences, Vol. 82, No. 6, 672 (1993)).
The structure formula of Argatroban is reported below:

- 21(S) Argatroban, X=CH3, Y═H;
- 21(R) Argatroban, X=H, Y=CH3;
- Argatroban, 21(S): 21 (R)=35:65.
The chemical names of the two diastereoisomers mentioned above are:
- 21(S) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3S)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-72-6); and
- 21(R) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3R)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-71-5).
In 1978, S. Akamoto et al from Japanese Mitsubishi Chemical Corporation first disclosed the anti-thrombin activity of Argatroban monohydrate (U.S. Pat. No. 4,101,653). In the next 20 years, numerous researchers had in-depth studies on Argatroban about its biological activity and medicine values. In 1981, S. Akamoto compared Argatroban with heparin in vivo (Okamoto, S. et al., Biochem. Biophys. Res. Commun. 101, 440 (1981)); T. Kumoto disclosed its three-dimensional selective activity (Kumada, T. et al., Thromb. Res. 24, 285 (1981)). In 1984, R. Kumato made a clinical evaluation of hemodialysis of Argatroban (Kikumoto, R. et al., Biochemistry 23, 85 (1984)), and in 1986, he further disclosed that Argatroban can inhibit the thrombin activity of mammals, and can be used as active ingredient to treat and prevent thrombosis and as an inhibitor of platelet aggregation. Argatroban monohydrate can be used as a selective anti-thrombosis agent for treatment of chronic arterial blockage and cerebral thrombosis, etc (JP 61-48829). In 1992 and 1993, Taparelli and Jakubowski separately disclosed the reversibility of Argatroban in anti-thrombin (Taparelli, C., Trends Pharmacol. Sci., 1993, 14, 366, Jakubowski, J. A. et al, Rep. Med. Chem., 1992, 27, 99). In 1990s, many researchers such as L. R. Buch reported other related research (Buch, L. R., Cadiosvasc. Drug Rev., 1991, 9, 247, Strupcnewski, J. D. et al., Academic: San Diego, 1991; Vol. 26, p 299, Brundish, D. et al., J. Med. Chem. 1999; 42, 4584, Shebuski, R. J., Academic: San Diego, 1999; Vol. 26, p 98). In 1992, Argatroban monohydrate was first approved as an anti-thrombin medicine in Japan (Hijikata-Okunomiya, A., et al, Thromb. Hemostasis, 1992, 18, 135).
Updated in sept 2015, reader there may be duplication
| Argatroban |
| AC1L99H9; AC-15185; TL8005144; C04931; | |
| MOLECULAR FORMULA: | C23H36N6O5S |
|---|---|
| MOLECULAR WEIGHT: | 508.63414 g/mol |
(2R,4R)-1-[5-(diaminomethylideneamino)-2-[(3-methyl-1,2,3,4-tetrahydroquinolin-8-yl)sulfonylamino]pentanoyl]-4-methylpiperidine-2-carboxylic acid, cas 74863-84-6
| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Prodrugs of (2R)-2-Propyloctanoic Acid For the Treatment of Stroke [US7495029] | 2008-06-05 | 2009-02-24 |
Tomiya Mano, Jin Shiomura, “Argatroban preparations for ophthalmic use.” U.S. Patent US5506241, issued October, 1986.
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in theliver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).
Argatroban is a direct, selective thrombin inhibitor. The American College of Cardiologists (ACC) recommend using bivalirudin or argatroban in patients who have had, or at risk for, heparin induced thrombocytopenia (HIT) and are undergoing percutaneous coronary intervention. Argatroban is a non-heparin anticoagulant shown to both normalize platelet count in patients with HIT and prevent the formation of thrombi. Parental anticoagulants must be stopped and a baseline activated partial thromboplastin time must be obtained prior to administering argatroban.

Transitioning to warfarin in individuals with heparin induced thrombocytopenia
Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.

http://www.google.com/patents/WO2009124906A2?cl=en
………….
NMR paper
Complete 1H and 13C assignments of (21R) and (21S) diastereomers of argatroban
Article first published online: 20 DEC 2007
DOI: 10.1002/mrc.2122, http://onlinelibrary.wiley.com/doi/10.1002/mrc.2122/abstract



click on image for clear view
1H NMR PREDICT


13C NMR PREDICT


References
1 Di Nisio M, Middeldorp S, Buller HR. Direct thrombin inhibitors. N Engl J Med 2005;353:1028-40. PMID 16148288
3Dhillon S. Argatroban: A Review of its Use in the Management of Heparin-Induced Thrombocytopenia. Am J Cardiovasc Drugs 2009; 9 (4): 261-82. Link text
- Hursting MJ, Lewis BE, Macfarlane DE. (2005). “Transitioning from argatroban to warfarin therapy in patients with heparin-induced thrombocytopenia.”. Clin Appl Thromb Hemost 11 (3): 279–87. doi:10.1177/107602960501100306. PMID 16015413.
External links
| EP0823430A1 * | Aug 4, 1997 | Feb 11, 1998 | Mitsubishi Chemical Corporation | Method for preparing n2-arylsulfonyl-l-argininamides |
| US4201863 * | Aug 31, 1978 | May 6, 1980 | Mitsubishi Chemical Industries, Limited | N2 -Arylsulfonyl-L-argininamides and the pharmaceutically acceptable salts thereof |
| REFERENCE | ||||
|---|---|---|---|---|
| 1 | None | |||
| 2 | * | OKAMOTO S ET AL: “Potent inhibition of thrombin by the newly synthesized arginine derivative No. 805. The importance of stereo-structure of its hydrophobic carboxamide portion” BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 101, no. 2, 30 July 1981 (1981-07-30), pages 440-446, XP024844713 ISSN: 0006-291X [retrieved on 1981-07-30] cited in the application | ||
| 3 | * | SONG H: “Method for preparing argatroban monohydrate in pure water” CASREACT,, 25 April 2007 (2007-04-25), XP002493272 & CN 1 951 937 A (TIANJIN WEIJIE TECHNOLOGY CO L [CN]) 25 April 2007 (2007-04-25) | ||
| CITING PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
| WO2012136504A1 | Mar 26, 2012 | Oct 11, 2012 | Lundbeck Pharmaceuticals Italy S.P.A. | Method for the preparation of process intermediates for the synthesis of argatroban monohydrate |
| CN102408468A * | Sep 20, 2011 | Apr 11, 2012 | 海南灵康制药有限公司 | Argatroban compound and preparation method thereof |
| EP2752412A1 | Mar 26, 2012 | Jul 9, 2014 | Lundbeck Pharmaceuticals Italy S.p.A. | Intermediates for the synthesis of Argatroban monohydrate |
Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5[(aminoiminomethyl)amino]1-oxo-2-[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4methyl-2-piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. The structural formula is:
|
Argatroban Injection is a sterile, non-pyrogenic, clear, colorless to pale yellow isotonic solution. It is supplied in a single use polyolefin bag containing 250 mg of argatroban in 250 mL sodium chloride solution (1 mg/mL). Each mL contains 1 mg argatroban, 9 mg sodium chloride, USP, and 3 mg sorbitol, NF in water for injection, USP. The pH of the solution is between 3.2 to 7.5.
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2- [[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl] sulfonylamino]pentanoyl]-4-methyl-piperidine-2- carboxylic acid | |
| CLINICAL DATA | |
| TRADE NAMES | Argatroban |
| AHFS/DRUGS.COM | monograph |
| ROUTES OF ADMINISTRATION | intravenous |
| PHARMACOKINETIC DATA | |
| BIOAVAILABILITY | 100% (intravenous) |
| PROTEIN BINDING | 54% |
| METABOLISM | hepatic |
| BIOLOGICAL HALF-LIFE | 39 and 51 minutes |
| IDENTIFIERS | |
| CAS REGISTRY NUMBER | 74863-84-6 |
| ATC CODE | B01AE03 |
| PUBCHEM | CID: 440542 |
| DRUGBANK | DB00278 |
| CHEMSPIDER | 389444 |
| UNII | OCY3U280Y3 |
| KEGG | C04931 |
| CHEMBL | CHEMBL1166 |
| CHEMICAL DATA | |
| FORMULA | C23H36N6O5S |
| MOLECULAR MASS | 508.635 g/mol |
//////



















