
CEP-1347; KT-7515
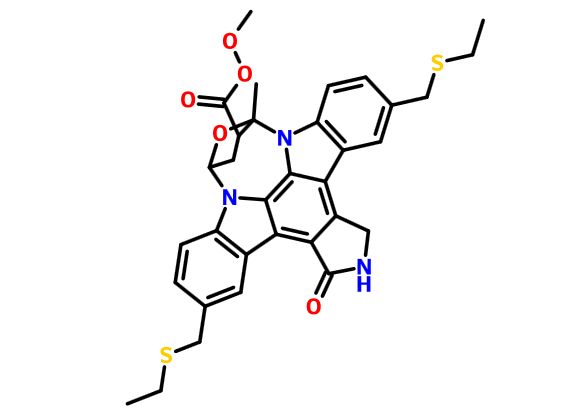
(9S,10R,12R)-5-16-Bis[(ethylthio)methyl]-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3',2',1'-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester
9,12-Epoxy-1H-diindolo(1,2,3-fg:3',2',1'-kl)pyrrolo(3,4-i)(1,6)benzodiazocine-10-carboxylic acid, 5,16-bis((ethylthio)methyl)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-, methyl ester, (9S-(9alpha,10beta,12alpha))-
METHYL (15S,16S,18S)-10,23-BIS[(ETHYLSULFANYL)METHYL]-15-METHYL-3-OXO-28-OXA-4,14,19-TRIAZAOCTACYCLO[12.11.2.1(1)?,(1)?.0(2),?.0?,(2)?.0?,(1)(3).0(1)?,(2)?.0(2)?,(2)?]OCTACOSA-1(26),2(6),7(27),8(13),9,11,20(25),21,23-NONAENE-16-CARBOPEROXOATE
A MAP3K11 (MLK3) inhibitor potentially for the treatment of Parkinson's disease.

9,12-Epoxy-1H-diindolo(1,2,3-fg:3',2',1'-kl)pyrrolo(3,4-i)(1,6)benzodiazocine-10-carboxylic acid, 5,16-bis((ethylthio)methyl)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-, methyl ester, (9S-(9alpha,10beta,12alpha))-
METHYL (15S,16S,18S)-10,23-BIS[(ETHYLSULFANYL)METHYL]-15-METHYL-3-OXO-28-OXA-4,14,19-TRIAZAOCTACYCLO[12.11.2.1(1)?,(1)?.0(2),?.0?,(2)?.0?,(1)(3).0(1)?,(2)?.0(2)?,(2)?]OCTACOSA-1(26),2(6),7(27),8(13),9,11,20(25),21,23-NONAENE-16-CARBOPEROXOATE
3,9-Bis(etsm)-K-252a; CEP1347; 3,9-Bis((ethylthio)methyl)-K-252a; AC1L31ZX
Phase III
A MAP3K11 (MLK3) inhibitor potentially for the treatment of Parkinson's disease.
MW 615.76, MF C33H33N3O5S2
Inhibitor of c-jun N-terminal kinase (JNK) signaling. Rescues motor neurons undergoing apoptosis (EC50 = 20 nM). Blocks Aβ-induced cortical neuron apoptosis (EC50 ~51 nM). Does not inhibit ERK1 activity. Neuroprotective.

Scheme 1 a
a (a) Ac2O, DMAP, THF, room temperature, 93%; (b) Cl2CHOCH3, TiCl4, CH2Cl2, 66%; (c) NaBH4 CH3OH, CHCl3, 65%; (d) NaOCH3, CH3OH, ClCH2CH2Cl, room temperature, 90%; (e) ROH, CSA, CH2Cl2; (f) RSH, CSA, CH2Cl2.
Inhibitor of c-jun N-terminal kinase (JNK) signaling. Rescues motor neurons undergoing apoptosis (EC50 = 20 nM). Blocks Aβ-induced cortical neuron apoptosis (EC50 ~51 nM). Does not inhibit ERK1 activity. Neuroprotective.
Apoptosis has been proposed as a mechanism of cell death in Alzheimer's, Huntington's and Parkinson's diseases and the occurrence of apoptosis in these disorders suggests a common mechanism.
Events such as oxidative stress, calcium toxicity, mitochondria defects, excitatory toxicity, and deficiency of survival factors are all postulated to play varying roles in the pathogenesis of the diseases.
However, the transcription factor c-jun may play a role in the pathology and cell death processes that occur in Alzheimer's disease.
Parkinson's disease (PD) is also a progressive disorder involving the specific degeneration and death of dopamine neurons in the nigrostriatal pathway. In Parkinson's disease, dopaminergic neurons in the substantia nigra are hypothesized to undergo cell death by apoptotic processes.
The commonality of biochemical events and pathways leading to cell death in these diseases continues to be an area under intense investigation.
The current therapy for PD and AD remains targeting replacement of lost transmitter, but the ultimate objective in neurodegenerative therapy is the functional restoration and/or cessation of progression of neuronal loss.
a novel approach for the treatment of neurodegenerative diseases through the development of kinase inhibitors that block the active cell death process at an early transcriptional independent step in the stress activated kinase cascade.
In particular, preclinical data will be presented on the c-Jun Amino Kinase pathway inhibitor, CEP-1347/KT-7515, with respect to it's properties that make it a desirable clinical candidate for treatment of various neurodegenerative diseases.
CEP-1347 is also known as KT-7515 and is being developed by Cephalon and Kyowa Hakko for treatment of Parkinson's disease and cognitive disorders.
It is believed to be a JNK-MAP kinase inhibitor. CEP-1347 has the chemical name 9alpha,12alpha-Epoxy-5,16-bis(ethylsulfanylmethyl)-10beta-hydroxy-9-methyl-1-oxo-2,3,9,10,11,12alpha-hexahydro-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4- i][1,6]benzodiazocine-10-carboxylic acid methyl ester and has the chemical structure as depicted in Formula 7.





PATENT
The compound with the structure outlined below is presently in clinical trials for Parkinson's disease (Idrugs, 2003, 6(4), 377-383).

This compound is in the following referred to as Compound I. The chemical name of Compound I is [9S-(9α,10β,12α)]-5,16-Rw[(ethylthio)methyl]-2,3,9,10,l l,12-hexahydro- 10-hydroxy-9-methyl- 1 -oxo-9, 12-epoxy- 1 H-diindolo[l ,2,3 -fg:3 ',2', 1 '-kl]ρyrrolo[3,4- i][l,6]benzodiazocine-10-carboxylic acid methyl ester.
The following references relate to Compound I, in particular to methods for its preparation [J.Med. Chem. 1997, 40(12), 1863-1869; Curr. Med. Chem. - Central Nervous System Agents, 2002, 2(2), 143-155] and its potential medical uses, mainly in diseases in the central nervous system (CNS), in particular for treatment of neurodegenerative diseases, e.g. Parkinson's disease, Alzheimer's disease, Huntington's disease, peripheral neuropathy, AIDS dementia, and ear injuries such as noise-induced hearing loss [Progress in Medicinal Chemistry (2002), 40, 23-62; Bioorg. Med. Chem. Lett. 2002,12(2), 147-150; Neuroscience, Oxford, 1998, 86(2), 461-472; J. Neurochemistry (2001), 77(3), 849-863; J. Neuroscience (2000), 20(1), 43-50; J. Neurochemistry (2002), 82(6), 1424-1434; Hearing Research, 2002, 166(1-2), 33-43].
The following patent documents relate to Compound I, including its medical use and synthesis: WO 9402488, WO9749406, US 5621100, EP 0651754 and EP 112 932. By the known methods, Compound I is synthesized in a solid amorphous form. The inventors have now discovered 5 crystalline forms of Compound I (named alpha, beta, gamma, delta and epsilon) thereby providing an opportunity to improve the manufacturing process of Compound I and its pharmaceutical use. There exists a need for crystalline forms, which may exhibit desirable and beneficial chemical and physical properties. There also exists a need for reliable and reproducible methods for the manufacture, purification, and formulation of Compound I to permit its feasible commercialisation.
EXAMPLES
In the following the starting material " Compound I" may, e.g., be prepared as described by Kaneko M. et al in J. Med. Chem. 1997, 40, 1863-1869.
Example 1. Preparation of crystalline alpha form of Compound I
Method I):
6.0 g amorphous Compound I was dissolved in 30 ml acetone. 0,6 g potassium carbonate was added and the suspension was stirred at room temperature for 1 hour before it was filtered to remove potential minor insoluble impurities and inorganic salts. The filter cake was washed with acetone. The filtrate was then evaporated on a rotary evaporator under reduced pressure at 60°C to a final volume of 10 ml to which 100 ml methanol was added slowly. The product separated as an oil, which almost dissolved on heating to reflux. Subsequently the residual insoluble impurities were removed by filtration. The filtrate was left with stirring at room temperature. A crystalline solid separated and was isolated by filtration. The filter cake was washed with methanol and dried in vacuo at 60°C overnight. Yield 2,83 g (47%), mp=182.4°C (DSC onset value), Weight loss by heating: 0.5%, Elemental analysis: 6.71%N, 63.93%C, 5.48%H, theoretical values corrected for 0.5% H2O: 6.79%N, 64.05%C, 5.43%H. XRPD analysis conforms with the alpha form. Method II):
5 g amorphous Compound I was dissolved in 25 ml acetone by gentle heating. 10 ml Methanol was added very slowly until the solution got turbid. The solution was allowed to cool to room temperature by natural cooling. The suspension was filtered and the filter-cake discarded. During filtration more material precipitated in the filtrate. The filtrate was heated until all material redissolves. Cold methanol was then added to the solution until precipitation was observed. The slightly turbid solution was then heated until all material was in solution. The solution was allowed to cool to room temperature, and the precipitate was removed by filtration. The second filter-cake was discarded. During the filtration some material separated in the filtrate. Heating redissolved the beginning crystallisation in the filtrate. Cold methanol was then added to the solution until precipitation was observed. The suspension was heated until a clear solution was obtained. The solution was allowed to reach room temperature by natural cooling. After a short period of time (15 min) precipitation begun. The precipitated pale yellow product was isolated by filtration and dried in vacuo at 50°C overnight. mp=188.9°C (DSC onset value), Weight loss by heating: 0.3%>, Elemental analysis: 6.53%N, 64.33%C, 5.43%H, theoretical values: 6.82%N, 64.37%C, 5.37%H. XRPD analysis conforms with the alpha form. Method III:
0.5g Compound I in a mixture of isopropyl acetate (10 mL) and water (0.6 mL) was heated to reflux with stirring. The compound was not completely dissolved so isopropyl acetate (10 mL) and water (0.6 mL) were added and heated to reflux. Stirring was stopped and the experiment was allowed to cool to room temperature. The crystalline product obtained were isolated by filtration and dried in vacuo at 40° C. Yield = 0.25g, mp = 183.7°C (DSC onset value). XRPD analysis conforms with the alpha form. Method IV: 0.5g Compound I in a mixture of ethyl acetate (10 mL) and water (0.4 mL) was heated to 70° C with stirring. The experiment was allowed to cool to room temperature. The crystalline product obtained were isolated by filtration and dried in vacuo at 40° C. XRPD analysis conforms with the alpha form.
PATENT
https://www.google.com/patents/US20050261762
PATENT
CEP-1347 (KT7515) (Maroney et al. 1998; Roux et al. 2002).

PAPER
Neurotrophic 3,9-bis[(alkylthio)methyl]- and -bis(alkoxymethyl)-K-252a derivatives
J Med Chem 1997, 40(12): 1863
J Med Chem 1997, 40(12): 1863


The synthesis of the title compound used as the starting material was the indolocarbazole alkaloid K-252A (I). Compound (I) was protected as the diacetyl derivative (II) by treatment with Ac2O and DMAP. Formylation of (II) with dichloromethyl methyl ether in the presence of TiCl4 afforded dialdehyde (III), which was further reduced to diol (IV) using NaBH4 in MeOH-CHCl3. Condensation of diol (IV) with ethanethiol in the presence of camphorsulfonic acid furnished the bis-sulfanyl compound (V). The acetyl protecting groups of (V) were finally removed by treatment with sodium methoxide. Alternatively, diol (IV) was first deacetylated by treatment with NaOMe, and the deprotected bis(hydroxymethyl) compound (VI) was then condensed with ethanethiol to produce the title bis-sulfayl compound 8.
3,9-Bis[(ethylthio)methyl]-K-252a (8):
mp 163−165 °C;
IR (KBr) 1725, 1680 cm-1; FAB-MSm/z 615(M+);
1H-NMR (400 MHz, DMSO-d6) δ 1.23 (t, 6H, J = 7.3 Hz), 1.99 (dd, 1H, J = 4.8, 14.1 Hz), 2.132 (s, 3H), 2.489 (q, 2H, J = 7.3 Hz), 2.505 (q, 2H, J = 7.3 Hz), 3.37 (dd, 1H, J = 7.6, 14.1 Hz), 3.92 (s, 3H), 3.94 (s, 2H), 3.98 (s, 2H), 4.95 (d, 1H, J = 17.6 Hz), 5.02 (d, 1H, J = 17.6 Hz), 6.32 (s, 1H), 7.10 (dd, 1H, J = 4.8, 7.6 Hz), 7.450 (m, 2H), 7.84 (d, 1H, J = 8.5 Hz), 7.88 (d, 1H, J = 8.8 Hz), 7.95 (d, 1H, J = 1.0 Hz), 8.60 (s, 1H), 9.13 (d, 1H, J = 0.7 Hz);
HRFAB-MS calcd for C33H33N3O5S2 615.1862, found 615.1869. Anal. (C33H33N3O5S2·0.5H2O) C, H, N.
References
Maroney et al (1998) Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J.Neurosci. 18 104. PMID: 9412490.
Saporito et al (1998) Preservation of cholinergic activity and prevention of neuron death by CEP-1347/KT-7515 following excitotoxic injury of the nucleus basalis magnocellularis. Neuroscience 86 461. PMID: 9881861.
Bozyczko-Coyne et al (2001) CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J.Neurochem. 77 849. PMID: 11331414.
| WO1994002488A1 * | Jul 26, 1993 | Feb 3, 1994 | Cephalon Inc | BIS-STAUROSPORINE AND K-252a DERIVATIVES |
| Reference | ||||
|---|---|---|---|---|
| 1 | * | KANEKO M ET AL: "Neurotrophic 3,9-Bis[(alkylthio)methyl]- and -Bis(alkoxymethyl)-K-252a Derivatives" JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 40, no. 12, 1997, pages 1863-1869, XP002128804 ISSN: 0022-2623 cited in the application | ||
//////////CEP 1347, KT 7515 ,
CCSCC1=CC2=C(C=C1)N3C4CC(C(O4)(N5C6=C(C=C(C=C6)CSCC)C7=C8CNC(=O)C8=C2C3=C75)C)C(=O)OOC




























