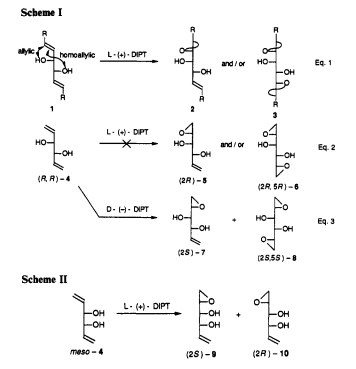
![ChemSpider 2D Image | 4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide | C45H50ClN7O7S](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_sArmFUFYbv9qowoxOw9HtjtHlRSsc5d6Kpo05oWMU97k9rmmNJxVt2e4lw3dn-qH0Ed49iuRhAgDfGc-TOTSAoDRCuFn-DUEV-qJycekrmuk1ILGobNKt83wsrZJ81vo0TvUY7HUo=s0-d)
ABT 199, RG 7601, GDC 0199
Venetoclax
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
SYNTHESIS UPDATED BELOW ..............

2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)-4-(4-((2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enyl)methyl)piperazin-1-yl)-N-(3-nitro-4-((tetrahydro-2H-pyran-4-yl)methylamino)phenylsulfonyl)benzamide
4-[4-[[2-(4-chlorophenyl)-4,4-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[3-nitro-4-(oxan-4-ylmethylamino)phenyl]sulfonyl-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
ABT 199
- Molecular Formula: C45H50ClN7O7S
- Average mass: 868.439209 Da
- Monoisotopic mass: 867.318115 Da
- 4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
NORTH CHICAGO, Ill., May 31, 2014/NEWS.GNOM.ES/ — AbbVie (NYSE: ABBV) released interim results from a Phase Ib clinical trial of ABT-199/GDC-0199, an investigational B-cell lymphoma 2 (BCL-2) selective inhibitor, in combination with rituximab (Abstract 7013). Results showed anoverall response rate (ORR) of 84 percent, in patients with relapsed/refractory chronic lymphocytic leukemia(CLL), the most common leukemia in the UnitedStates. These results were presented at the 50thAnnual Meeting of the American Society of ClinicalOncology (ASCO), May 30 – June 3 in Chicago.
ABT-199 is a so-called BH3-mimetic drug, which is designed to block the function of the protein Bcl 2. In 1988, it was discovered that Bcl-2 allowed leukaemia cells to become long-lived, a discovery made at the Walter and Eliza Hall Institute by Professors David Vaux, Suzanne Cory and Jerry Adams. Subsequent research led by them and other institute scientists, including Professors Andreas Strasser, David Huang, Peter Colman and Keith Watson, has explained much about how Bcl-2 and related molecules function to determine if a cell lives or dies. These discoveries have contributed to the development of a new class of drugs called BH3-mimetics that kill, and thereby rapidly remove, leukaemic cells by blocking Bcl-2. (source:http://www.wehi.edu.au).
Highlights of recent research using this agent
|
GDC-0199 (RG7601) is a novel small molecule Bcl-2 selective inhibitor designed to restore apoptosis, also known as programmed cell death, by blocking the function of a pro-survival Bcl-2 family protein. The Bcl-2 family proteins, which are expressed at high levels in many tumors, play a central role in regulating apoptosis and, consequently, are thought to impact tumor formation, tumor growth and resistance.
Venetoclax (previously: GDC-0199, ABT-199, RG7601 )[1] is a small molecule oral drug being investigated to treat chronic lymphocytic leukemia (CLL).[2][3]
In 2015, the FDA granted Breakthrough Therapy Designation to venetoclax for CLL in previously treated (relapsed/refractory) patients with the 17p deletion genetic mutation.[3]
Mechanism of action
Venetoclax (a BH3-mimetic[4]) acts as a Bcl-2 inhibitor, ie. it blocks the anti-apoptotic B-cell lymphoma-2 (BCL2) protein, leading toprogrammed cell death in CLL cells.[2]
Clinical trials
A phase 1 trial established a dose of 400mg/day.[2]
A phase 2 trial met its primary endpoint which was overall response rate.[3] Interim results from a Phase 2b trial are encouraging, especially in patients with the 17p deletion.[2]
NOW IN PHASE 3 UPDATED............

4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
(hereafter, “Compound 1”) is a potent and selective Bcl-2 inhibitor
having, inter alia, antitumor activity as an apoptosis-inducing agent.
Compound 1 has the formula:

Compound
1 is currently the subject of ongoing clinical trials for the treatment
of chronic lymphocytic leukemia. U.S. Patent Publication No.
2010/0305122 describes Compound 1, and other compounds which exhibit
potent binding to a Bcl-2 family protein, and pharmaceutically
acceptable salts thereof. U.S. Patent Publication Nos. 2012/0108590 and
2012/0277210 describe pharmaceutical compositions comprising such
compounds, and methods for the treatment of neoplastic, immune or
autoimmune diseases comprising these compounds. U.S. Patent Publication
No. 2012/0157470 describes pharmaceutically acceptable salts and
crystalline forms of Compound 1. The disclosures of U.S. 2010/0305122;
2012/0108590; 2012/0157470 and 2012/0277210 are hereby incorporated by
reference in their entireties.



PATENT
US 2015183783
PATENT
CN 104370905
http://www.google.com/patents/CN104370905A?cl=en
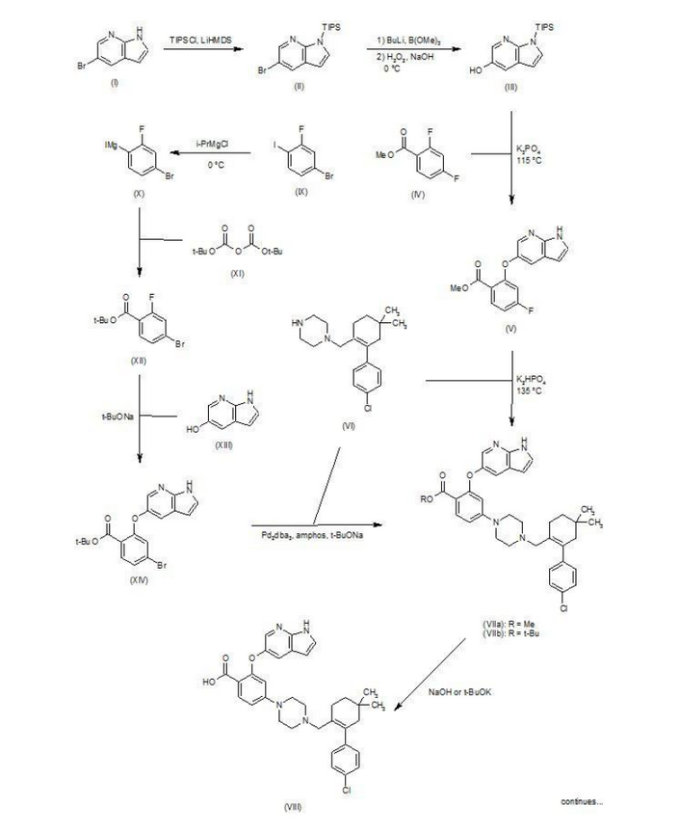
ABT-199 is developed AbbVie Bel-2 inhibitors, I trial (NCT01328626) enrolled 84 patients with relapsed type / refractory CLL / SLL patients and 44 cases of relapsing / refractory non-Hodgkin lymphoma patients. ABT-199 treatment response CLL / SLL rate of 79% (complete response rate of 22%), median duration of response time was 20.5 months; ABT-199 treatment of non-Hodgkin's lymphoma response rate of 48% (complete response rate was 7.5%). The efficacy of ABT-199 is capable of obinutuzumab, idelalisib, ibrutinib rival, is expected to become the first listed Bcl_2 inhibitors, ABT-199 is currently ongoing Phase III clinical study.
ABT-199 compound CAS number 1257044-40-8, the compound is structured as follows:
 Patent W02012058392, W02012071336, W02010138588 et al. Discloses the
preparation of ABT-199 in order to -IH- 5-bromo-pyrrolo [2, 3-b]
pyridine as raw material to protect hydroxylation, after replacing the
compound 5, and reaction of compound 6, hydrolysis to give compound 9,
compound 10 and compound 9 obtained by condensation of ABT-199, a
specific line as follows:
Patent W02012058392, W02012071336, W02010138588 et al. Discloses the
preparation of ABT-199 in order to -IH- 5-bromo-pyrrolo [2, 3-b]
pyridine as raw material to protect hydroxylation, after replacing the
compound 5, and reaction of compound 6, hydrolysis to give compound 9,
compound 10 and compound 9 obtained by condensation of ABT-199, a
specific line as follows:


use
of 2-fluoro-4-nitrobenzoate (A) as a raw material, and substituted
5-hydroxy-7-aza-indole (B), reduction to produce compound ( D), the
compound (D) with the compound by cyclization after (H) substitution,
hydrolysis to yield compound (J), and then with the compound (K) to
afford ABT-199.
Preparation of a compound of Example (F) of the
Example
 First step: Synthesis of Compound (C)
First step: Synthesis of Compound (C)
2-fluoro-4-nitrobenzoate in IL three-necked flask 50. 0g, dissolved with dimethylformamide N'N- 250ml, was added successively 5-hydroxy-7-aza-indole indole 33. 6g, potassium carbonate 34. 7g, the reaction was heated to 50 degrees under nitrogen gas protection for 2 hours, poured into 2L of ice water was added and extracted three times with ethyl acetate, the organic phase was dried with saturated sodium chloride spin dry to give Compound (C) crude 82. 0g, crude without purification in the next reaction direct investment.
Step two: Synthesis of Compound (D)
The compound of the previous step (C) of the crude product was dissolved in methanol 400ml, was added 10% palladium on carbon 4. 0g, through the reaction of hydrogen at atmospheric pressure, after the end of the reaction by TLC spin solvent to give compound (D) The crude product 73. 2g, crude without purification in the next reaction direct investment.
The third step: Synthesis of compound (F)
Take the previous step the compound (D) crude 20. 0g, t-butanol were added 150ml, compound (E) 10. g, potassium carbonate 9. 7g, completion of the addition the reaction was refluxed for 48 hours the reaction solution was cooled, added acetic acid ethyl ester was diluted, washed with water three times, the combined aqueous phases extracted once with ethyl acetate, the combined ethyl acetate phases twice, dried over anhydrous sodium sulfate and the solvent was spin, the crude product obtained was purified by silica gel column chromatography to give 13. 9g, three-step overall yield of 57.4%.
Preparation Example II Compound (H),
 [0029] Take compound (G) (prepared according to W02012058392 method) 5.
0g, dissolved with 50ml of dichloromethane, was added triethylamine 5.
6ml, the reaction solution was cooled to 0-5 ° with stirring, was added
dropwise methanesulfonyl chloride 2. 7g, the addition was complete the
reaction was warmed to room temperature overnight, after the end of the
reaction by TLC the reaction was quenched with water, the organic phase
was dried over anhydrous sodium sulfate and the solvent was spin,
purified by silica gel column chromatography to give compound (H) 6. 5g ,
a yield of 99%.
[0029] Take compound (G) (prepared according to W02012058392 method) 5.
0g, dissolved with 50ml of dichloromethane, was added triethylamine 5.
6ml, the reaction solution was cooled to 0-5 ° with stirring, was added
dropwise methanesulfonyl chloride 2. 7g, the addition was complete the
reaction was warmed to room temperature overnight, after the end of the
reaction by TLC the reaction was quenched with water, the organic phase
was dried over anhydrous sodium sulfate and the solvent was spin,
purified by silica gel column chromatography to give compound (H) 6. 5g ,
a yield of 99%.
Three ABT-199 Preparation of Example
 First step: Synthesis of Compound (I)
First step: Synthesis of Compound (I)
In IOOml three-necked flask were added the compound (F) 2. 5g, compound ⑶2. 3g, potassium carbonate I. 9g, Ν 'was added and reacted at 50 degrees N- dimethylformamide 15ml, nitrogen atmosphere, TLC detection After the reaction, the reaction solution was poured into ice-water, extracted with ethyl acetate twice added ethyl acetate phase was dried over anhydrous sodium sulfate spin, and purified by silica gel column chromatography to give compound (I) 3. 6g, yield 88 %.
Step two: Synthesis of Compound (J)
In IOml single jar Compound (I) I. 0g, followed by adding water 5ml, ethanol 5ml, tetrahydrofuran 5ml, sodium hydroxide 136mg, the reaction was stirred at room temperature the reaction, ethyl acetate was added after dilution of the reaction by TLC, adjusted with IN hydrochloric acid PH4-5, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate and spin dried to give compound (J) 907mg, 93% yield.
Step two: Synthesis of ABT-199
In a 25ml single neck flask was added the compound (J) 100mg, EDCI67mg, dichloromethane 10ml, the reaction was stirred for 30 minutes, was added the compound (K) (prepared in accordance with W02012058392) 55mg, finally added a catalytic amount of DMAP, the force After opening the reaction was stirred overnight, after the end of the reaction by TLC the solvent was spin, HPLC purified preparation obtained by pure ABT-199 ^ 9811, 65% yield.
ABT-199 is developed AbbVie Bel-2 inhibitors, I trial (NCT01328626) enrolled 84 patients with relapsed type / refractory CLL / SLL patients and 44 cases of relapsing / refractory non-Hodgkin lymphoma patients. ABT-199 treatment response CLL / SLL rate of 79% (complete response rate of 22%), median duration of response time was 20.5 months; ABT-199 treatment of non-Hodgkin's lymphoma response rate of 48% (complete response rate was 7.5%). The efficacy of ABT-199 is capable of obinutuzumab, idelalisib, ibrutinib rival, is expected to become the first listed Bcl_2 inhibitors, ABT-199 is currently ongoing Phase III clinical study.
ABT-199 compound CAS number 1257044-40-8, the compound is structured as follows:

Preparation of a compound of Example (F) of the
Example

2-fluoro-4-nitrobenzoate in IL three-necked flask 50. 0g, dissolved with dimethylformamide N'N- 250ml, was added successively 5-hydroxy-7-aza-indole indole 33. 6g, potassium carbonate 34. 7g, the reaction was heated to 50 degrees under nitrogen gas protection for 2 hours, poured into 2L of ice water was added and extracted three times with ethyl acetate, the organic phase was dried with saturated sodium chloride spin dry to give Compound (C) crude 82. 0g, crude without purification in the next reaction direct investment.
Step two: Synthesis of Compound (D)
The compound of the previous step (C) of the crude product was dissolved in methanol 400ml, was added 10% palladium on carbon 4. 0g, through the reaction of hydrogen at atmospheric pressure, after the end of the reaction by TLC spin solvent to give compound (D) The crude product 73. 2g, crude without purification in the next reaction direct investment.
The third step: Synthesis of compound (F)
Take the previous step the compound (D) crude 20. 0g, t-butanol were added 150ml, compound (E) 10. g, potassium carbonate 9. 7g, completion of the addition the reaction was refluxed for 48 hours the reaction solution was cooled, added acetic acid ethyl ester was diluted, washed with water three times, the combined aqueous phases extracted once with ethyl acetate, the combined ethyl acetate phases twice, dried over anhydrous sodium sulfate and the solvent was spin, the crude product obtained was purified by silica gel column chromatography to give 13. 9g, three-step overall yield of 57.4%.
Preparation Example II Compound (H),

Three ABT-199 Preparation of Example

In IOOml three-necked flask were added the compound (F) 2. 5g, compound ⑶2. 3g, potassium carbonate I. 9g, Ν 'was added and reacted at 50 degrees N- dimethylformamide 15ml, nitrogen atmosphere, TLC detection After the reaction, the reaction solution was poured into ice-water, extracted with ethyl acetate twice added ethyl acetate phase was dried over anhydrous sodium sulfate spin, and purified by silica gel column chromatography to give compound (I) 3. 6g, yield 88 %.
Step two: Synthesis of Compound (J)
In IOml single jar Compound (I) I. 0g, followed by adding water 5ml, ethanol 5ml, tetrahydrofuran 5ml, sodium hydroxide 136mg, the reaction was stirred at room temperature the reaction, ethyl acetate was added after dilution of the reaction by TLC, adjusted with IN hydrochloric acid PH4-5, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate and spin dried to give compound (J) 907mg, 93% yield.
Step two: Synthesis of ABT-199
In a 25ml single neck flask was added the compound (J) 100mg, EDCI67mg, dichloromethane 10ml, the reaction was stirred for 30 minutes, was added the compound (K) (prepared in accordance with W02012058392) 55mg, finally added a catalytic amount of DMAP, the force After opening the reaction was stirred overnight, after the end of the reaction by TLC the solvent was spin, HPLC purified preparation obtained by pure ABT-199 ^ 9811, 65% yield.
PATENT
WO 2014165044
PATENT
US 2014275540
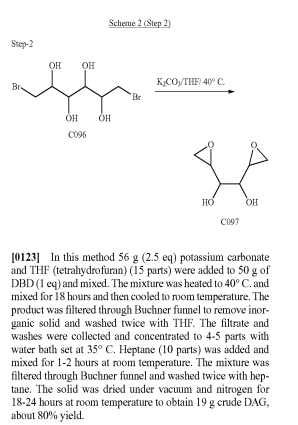
http://www.google.com/patents/US20140275540

- An exemplary reaction according to Scheme 2 is shown below.Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302.
- Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218.
- In another embodiment, the compound of formula (1) is prepared from compound (D) and compound (I) as shown in Scheme 5 below. Compound (J) may be prepared by techniques known in the art, e.g., as shown in WO 2009/117626 and Organometallics, 2008, 27 (21), 5605-5611.
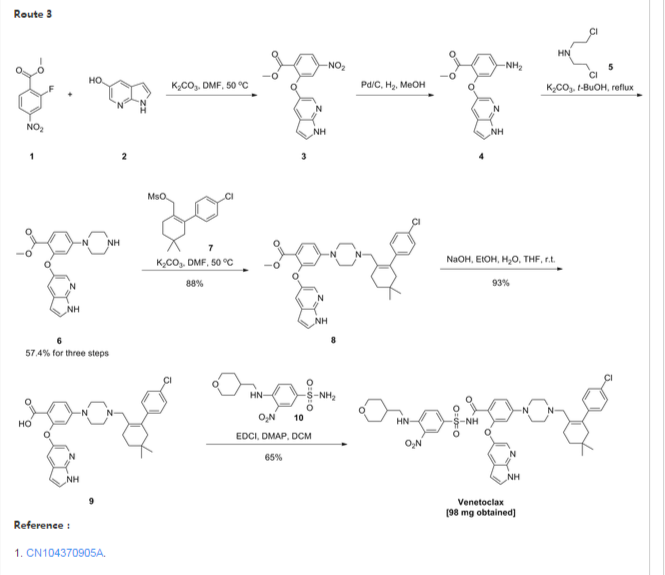
- Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H).
- Example 2 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-bromobenzoate (Compound (D))
- To a 3 L three-neck Morton flask were charged 1H-pyrrolo[2,3-b]pyridin-5-ol (80.0 g, 1.00 eq.), tert-butyl 4-bromo-2-fluorobenzoate (193 g, 1.15 eq.), and anhydrous DMF (800 mL). The mixture was stirred at 20° C. for 15 min. The resulting solution was cooled to about zero to 5° C. A solution of sodium tert-butoxide (62.0 g) in DMF (420 mL) was added slowly over 30 min while maintaining the internal temperature at NMT 10° C., and rinsed with DMF (30 mL). The reaction mixture was stirred at 10° C. for 1 hour (an off-white slurry) and adjusted the internal temperature to ˜45° C. over 30 min. The reaction mixture was stirred at 45-50° C. for 7 hr and the reaction progress monitored by HPLC (IP samples: 92% conversion % by HPLC). The solution was cooled to ˜20° C. The solution was stirred at 20° C. overnight.
- Water (1200 mL) was added slowly to the reaction mixture at <30° C. over 1 hour (slightly exothermic). The product slurry was adjusted to ˜20° C., and mixed for NLT 2 hours. The crude product was collected by filtration, and washed with water (400 mL). The wet-cake was washed with heptane (400 mL) and dried under vacuum at 50° C. overnight to give the crude product (236.7 g).
- Re-crystallization or Re-slurry: 230.7 g of the crude product, (potency adjusted: 200.7 g) was charged back to a 3 L three-neck Morton flask. Ethyl acetate (700 mL) was added, and the slurry heated slowly to refluxing temperature over 1 hr (small amount of solids left). Heptane (1400 mL) was added slowly, and the mixture adjusted to refluxing temperature (78° C.). The slurry was mixed at refluxing temperature for 30 min., and cooled down slowly to down to ˜−10° C. at a rate of approximate 10° C./hour), and mixed for 2 hr. The product was collected by filtration, and rinsed with heptane (200 ml).
- The solid was dried under vacuum at ˜50° C. overnight to give 194.8 g, 86% isolated yield of the product as an off-white solid. MS-ESI 389.0 (M+1); mp: 190-191° C. (uncorrected). 1H NMR (DMSO-d6): δ 1.40 (s, 9H), 6.41 (dd, J=3.4, 1.7 Hz, 1H), 7.06 (d, J=1.8 Hz, 1H), 7.40 (dd, J=8.3, 1.8 Hz, 1H), 7.51 (t, J=3.4 Hz, 1H), 7.58 (d, J=2.6 Hz, 1H), 7.66 (d, J=8.3 Hz, 1H), 8.03 (d, J=2.7 Hz, 1H), 11.72 (s, 1H, NH).
- Example 3 Synthesis of 2-chloro-4,4-dimethylcyclohexanecarbaldehyde (Compound (F))
- To a 500 mL RB flask were charged anhydrous DMF (33.4 g, 0.456 mol) and CH2Cl2 (80 mL). The solution was cooled down <−5° C., and POCl3 (64.7 g, 0.422 mol) added slowly over 20 min @<20° C. (exothermic), rinsed with CH2Cl2 (6 mL). The slightly brown solution was adjusted to 20° C. over 30 min, and mixed at 20° C. for 1 hour. The solution was cooled back to <5° C. 3,3-Dimethylcyclohexanone (41.0 g, 90%, ˜0.292 mol) was added, and rinsed with in CH2Cl2 (10 mL) (slightly exothermic) at <20° C. The solution was heated to refluxing temperature, and mixed overnight (21 hours).
- To a 1000 mL three neck RB flask provided with a mechanical stirrer were charged 130 g of 13.6 wt % sodium acetate trihydrate aqueous solution, 130 g of 12% brine, and 130 mL of CH2Cl2. The mixture was stirred and cooled down to <5° C. The above reaction mixture (clear and brown) was transferred, quenched into it slowly while maintaining the internal temperature <10° C. The reaction vessel was rinsed with CH2Cl2 (10 mL). The quenched reaction mixture was stirred at <10° C. for 15 min. and allowed to rise to 20° C. The mixture was stirred 20° C. for 15 min and allowed to settle for 30 min. (some emulsion). The lower organic phase was separated. The upper aq. phase was back extracted with CH2Cl2 (50 mL). The combined organic was washed with a mixture of 12% brine (150 g)-20% K3PO4 aq. solution (40 g). The organic was dried over MgSO4, filtered and rinsed with CH2Cl2 (30 ml). The filtrate was concentrated to dryness under vacuum to give a brown oil (57.0 g, potency=90.9 wt % by qNMR, ˜100%). 1H NMR (CDCl3): δ 0.98 (s, 6H), 1.43 (t, J=6.4 Hz, 2H), 2.31 (tt, J=6.4, 2.2 Hz, 2H), 2.36 (t, J=2.2 Hz, 2H), 10.19 (s, 1H).
- Example 4 Synthesis of 2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enecarbaldehyde (Compound (G))
- To a 250 mL pressure bottle were charged 2-chloro-4,4-dimethylcyclohex-1-enecarbaldehyde (10.00 g), tetrabutylammonium bromide (18.67 g), and acetonitrile (10 mL). The mixture was stirred at 20° C. for 5 min. 21.0 wt % K2CO3 aq. solution (76.0 g) was added. The mixture was stirred at room temperature (rt) for NLT 5 min. followed by addition of 4-chlorophenylboronic acid (9.53 g) all at once. The mixture was evacuated and purged with N2 for three times. Palladium acetate (66 mg, 0.5 mol %) was added all at once under N2. The reaction mixture was evacuated and purged with N2 for three times (an orange colored mixture). The bottle was back filled with N2 and heated to ˜35° C. in an oil bath (bath temp ˜35° C.). The mixture was stirred at 30° C. overnight (15 hours). The reaction mixture was cooled to RT, and pulled IP sample from the upper organic phase for reaction completion, typically starting material <2% (orange colored mixture). Toluene (100 mL) and 5% NaHCO3-2% L-Cysteine aq. solution (100 mL) were added. The mixture was stirred at 20° C. for 60 min. The mixture was filtered through a pad of Celite to remove black solid, rinsing the flask and pad with toluene (10 mL). The upper organic phase was washed with 5% NaHCO3 aq. solution-2% L-Cysteine (100 mL) once more. The upper organic phase was washed with 25% brine (100 mL). The organic layer (105.0 g) was assayed (118.8 mg/g, 12.47 g product assayed, 87% assayed yield), and concentrated to ˜1/3 volume (˜35 mL). The product solution was directly used in the next step without isolation. However, an analytical sample was obtained by removal of solvent to give a brown oil. 1HNMR (CDCl3): δ 1.00 (s, 6H), 1.49 (t, J=6.6 Hz, 2H), 2.28 (t, J=2.1 Hz, 2H), 2.38 (m, 2H), 7.13 (m, 2H), 7.34 (m, 2H), 9.47 (s, 1H).
- Example 5 Synthesis of tert-butyl 4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine-1-carboxylate (Compound (H))
- To a 2 L three neck RB flask provided with a mechanical stirrer were charged a solution of 4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-carbaldehyde (50.0 g) in toluene (250 mL), BOC-piperazine (48.2 g) and anhydrous THF (250 mL). The yellow solution was stirred at 20° C. for 5 min. Sodium triacetoxyborohydride (52.7 g) was added in portion (note: the internal temperature rose to ˜29.5° C. in 15 min cooling may be needed). The yellow mixture was stirred at ˜25° C. for NLT 4 hrs. A conversion of starting material to product of 99.5% was observed by HPLC after a 3 hour reaction time.
- 12.5 wt % brine (500 g) was added slowly to quench the reaction. The mixture was stirred at 20° C. for NLT 30 min and allowed to settle for NLT 15 min. The lower aq. phase (˜560 mL) was separated (note: leave any emulsion in the upper organic phase). The organic phase was washed with 10% citric acid solution (500 g×2). 500 g of 5% NaHCO3 aq. solution was charged slowly into the flask. The mixture was stirred at 20° C. for NLT 30 min., and allowed to settle for NLT 15 min. The upper organic phase was separated. 500 g of 25% brine aq. solution was charged. The mixture was stirred at 20° C. for NLT 15 min., and allowed to settle for NLT 15 min. The upper organic phase was concentrated to ˜200 mL volume under vacuum. The solution was adjusted to −30° C., and filtered off the inorganic salt. Toluene (50 mL) was used as a rinse. The combined filtrate was concentrated to ˜100 mL volume. Acetonitrile (400 mL) was added, and the mixture heated to ˜80° C. to achieve a clear solution. The solution was cooled down slowly to 20° C. slowly at rate 10° C./hour, and mixed at 20° C. overnight (the product is crystallized out at ˜45-50° C., if needed, seed material may be added at 50° C.). The slurry was continued to cool down slowly to ˜−10° C. at rate of 10° C./hours. The slurry was mixed at ˜−10° C. for NLT 6 hours. The product was collected by filtration, and rinsed with pre-cooled acetonitrile (100 mL). The solid was dried under vacuum at 50° C. overnight (72.0 g, 85%). MS-ESI: 419 (M+1); mp: 109-110° C. (uncorrected); 1H NMR (CDCl3): δ 1.00 (s, 6H), 1.46 (s, 9H), 1.48 (t, J=6.5 Hz, 2H), 2.07 (s, br, 2H), 2.18 (m, 4H), 2.24 (t, J=6.4 Hz, 2H), 2.80 (s, 2H), 3.38 (m, 4H), 6.98 (m, 2H), 7.29 (m, 2H).
- Example 6 Synthesis of 1-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine dihydrochloride (Compound (I))
- To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the Boc reductive amination product (Compound (H), 72.0 g) and IPA (720 mL). The mixture was stirred at rt for 5 min, and 59.3 g of concentrated hydrochloride aq. solution added to the slurry. The reaction mixture was adjusted to an internal temperature of ˜65° C. (a clear and colorless solution achieved). The reaction mixture was agitated at ˜65° C. for NLT 12 hours.
- The product slurry was cooled down to −5° C. slowly (10° C./hour). The product slurry was mixed at ˜−5° C. for NLT 2 hours, collected by filtration. The wet cake was washed with IPA (72 mL) and dried at 50° C. under vacuum overnight to give 73.8 g (95%) of the desired product as a bis-hydrochloride IPA solvate (purity >99.5% peak area at 210 nm). MS-ESI: 319 (M+1); 1HNMR (CDCl3): δ 0.86 (s, 6H), 1.05 (d, J=6.0 Hz, 6H, IPA), 1.42 (t, J=6.1 Hz, 2H), 2.02 (s, br, 2H), 2.12 (m, 2H), 3.23 (m, 4H), 3.4 (s, br, 4H), 3.68 (s, 2H), 3.89 (septet, J=6.0 Hz, 1H, IPA), 7.11 (d, J=8.1 Hz, 2H), 7.41 (d, J=8.1 Hz, 2H).
- Example 7 Synthesis of 3-nitro-4-(((tetrahydro-2H-pyran-4-yl)methyl)amino)-benzenesulfonamide (Compound (N))
- To a 500 mL three-neck RB flask equipped with a mechanical stirrer were charged the 4-chloro-3-nitrobenzenesulfonamide, Compound M (10.0 g), diisopropylethylamine (17.5 g), (tetrahydro-2H-pyran-4-yl)methanamine (7.0 g) and acetonitrile (150 mL). The reaction mixture was adjusted to an internal temperature of 80° C. and agitated for no less than 12 hours.
- The product solution was cooled down to 40° C. and agitated for no less than 1 hour until precipitation observed. The product slurry was further cooled to 20° C. Water (75 mL) was slowly charged over no less than 1 hour, and the mixture cooled to 10° C. and agitated for no less than 2 hours before collected by filtration. The wet cake was washed with 1:1 mix of acetonitrile:water (40 mL). The wet cake was then reslurried in water (80 mL) at 40° C. for no less than 1 hour before collected by filtration. The wet cake was rinsed with water (20 mL), and dried at 75° C. under vacuum to give 12.7 g of the desired product in 99.9% purity and in 91% weight-adjusted yield. 1H NMR (DMSO-d6): δ 1.25 (m, 2H), 1.60 (m, 2H), 1.89 (m, 1H), 3.25 (m, 2H), 3.33 (m, 2H), 3.83 (m, 2H), 7.27 (d, J=9.3 Hz, 1H), 7.32 (s, NH2, 2H), 7.81 (dd, J=9.1, 2.3 Hz, 1H), 8.45 (d, J=2.2 Hz, 1H), 8.54 (t, J=5.9 Hz, 1H, NH).
- Example 8 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate (Compound (K))
- General Considerations:
- this chemistry is considered air and moisture sensitive. While the catalyst precursors in their solid, dry form can be handled and stored in air without special precautions, contact with even small amounts of solvent may render them susceptible to decomposition. As a result, traces of oxygen or other competent oxidants (e.g., solvent peroxides) must be removed prior to combination of the catalyst precursors with solvent and care must be used to prevent ingress of oxygen during the reaction. Also, care must be taken to use dry equipment, solvents, and reagents to prevent formation of undesirable byproducts. The sodium t-butoxide used in this reaction is hygroscopic and it should be properly handled and stored prior to or during use.
- To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the bis-hydrochloride salt (Compound (I), 42.5 g) and toluene (285 ml). 20% K3PO4 (285 ml) was added and the biphasic mixture was stirred for 30 min. The layers were separated and the organic layer was washed with 25% NaCl (145 ml). The organic layer concentrated to 120 g and used in the coupling reaction without further purification.
- NaOtBu (45.2 g) and Compound (I) in toluene solution (120 g solution −30 g potency adjusted) were combined in THF (180 ml) in a suitable reactor and sparged with nitrogen for NLT 45 min. Pd2dba3 (0.646 g), Compound (J) (0.399 g), and Compound (D) (40.3 g) were combined in a second suitable reactor and purged with nitrogen until oxygen level was NMT 40 ppm. Using nitrogen pressure, the solution containing Compound (I) and NaOtBu in toluene/THF was added through a 0.45 μm inline filter to the second reactor (catalyst, Compound (J) and Compound (D)) and rinsed with nitrogen sparged THF (30 ml).
- The resulting mixture was heated to 55° C. with stirring for NLT 16 h, then cooled to 22° C. The mixture was diluted with 12% NaCl (300 g) followed by THF (300 ml). The layers were separated.
- The organic layer was stirred with a freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated.
- The organic layer was stirred with a second freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated. The organic layer was washed with 12% NaCl (300 g), then filtered through a 0.45 μm inline filter. The filtered solution was concentrated in vacuo to ˜300 mL, and chased three times with heptane (600 mL each) to remove THF.
- The crude mixture was concentrated to 6 volumes and diluted with cyclohexane (720 ml). The mixture was heated to 75° C., held for 15 min, and then cooled to 65° C. over NLT 15 min. Seed material was charged and the mixture was held at 65° C. for 4 hours. The suspension was cooled to 25° C. over NLT 8 h, then held at 25° C. for 4 hours. The solids were filtered and washed with cyclohexane (90 ml) and dried at 50° C. under vacuum.
- Isolated 52.5 g (88.9% yield) as a white solid. Melting point (uncorrected) 154-155° C. 1H NMR (DMSO-d6): δ 0.93 (s, 6H), 1.27 (s, 9H), 1.38 (t, J=6.4 Hz, 2H), 1.94 (s, 2H), 2.08-2.28 (m, 6H), 2.74 (s, 2H), 3.02-3.19 (m, 4H), 6.33 (dd, J=3.4, 1.9 Hz, 1H), 6.38 (d, J=2.4 Hz, 1H), 6.72 (dd, J=9.0, 2.4 Hz, 1H), 6.99-7.06 (m, 2H), 7.29 (d, J=2.7 Hz, 1H), 7.30-7.36 (m, 2H), 7.41-7.44 (m, 1H), 7.64 (t, J=6.7 Hz, 1H), 7.94 (d, J=2.7 Hz, 1H), 11.53 (s, 1H).
- Example 9 Synthesis of 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoic acid (Compound (L))
- Solution preparation: 10% KH2PO4 (aq): KH2PO4 (6 g) in water (56 g); 2:1 heptane/2-MeTHF:heptane (16 mL) in 2-MeTHF (8 mL).
- Compound (K) (5.79 g), potassium tert-butoxide (4.89 g), 2-methyltetrahydrofuran (87 mL), and water (0.45 mL) were combined in a suitable reactor under nitrogen and heated to 55° C. until reaction completion. The reaction mixture was cooled to 22° C., washed with the 10% KH2PO4 solution (31 g) twice. The organic layer was then washed with water (30 g).
- After removal of the aqueous layer, the organic layer was concentrated to 4 volumes (˜19 mL) and heated to no less than 50° C. Heptane (23 ml) was slowly added. The resulting suspension was cooled to 10° C. Solids were then collected by vacuum filtration with recirculation of the liquors and the filter cake washed with 2:1 heptane/2-MeTHF (24 ml). Drying of the solids at 80° C. under vacuum yielded 4.0 g of Compound (L) in approximately 85% weight-adjusted yield. 1H NMR (DMSO-d6): δ 0.91 (s, 6H), 1.37 (t, J=6.4 Hz, 2H), 1.94 (s, br, 2H), 2.15 (m, 6H), 2.71 (s, br, 2H), 3.09 (m, 4H), 6.31 (d, J=2.3 Hz, 1H), 6.34 (dd, J=3.4, 1.9 Hz, 1H), 6.7 (dd, J=9.0, 2.4 Hz, 1H), 7.02 (m, 2H), 7.32 (m, 2H), 7.37 (d, J=2.6 Hz, 1H), 7.44 (t, J=3.0 Hz, 1H), 7.72 (d, J=9.0 Hz, 1H), 7.96 (d, J=2.7 Hz, 1H) & 11.59 (m, 1H).
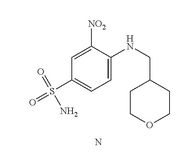
- Example 10 Synthesis of 4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (Compound (I))
- Solution preparation prior to reaction: 10% Acetic Acid:Acetic Acid (37 mL) in water (333 g); 5% NaHCO3:NaHCO3 (9 g) in water (176 g); 5% NaCl:NaCl (9 g) in water (176 g).
- Compound (N) (13.5 g), DMAP (10.5 g), EDAC (10.7 g) and dichloromethane (300 mL) were combined in a suitable reactor and agitated at 25° C. In a second suitable reactor was charged the Acid (Compound (L), 25 g), Et3N (8.7 g) and dichloromethane (120 mL). The resulting Acid (Compound (L)) solution was slowly charged to the initial suspension of Compound (N) and agitated until reaction completion.
 COMPD L
COMPD L
 COMPD N
COMPD N
- 1H NMR (DMSO-d6): δ 0.90 (s, 6H), 1.24 (m, 2H), 1.36 (t, J=6.4 Hz, 2H), 1.60 (m, 2H), 1.87 (m, 1H), 1.93 (s, br, 2H), 2.12 (m, 2H), 2.19 (m, 4H), 2.74 (s, br, 2H), 3.06 (m, 4H), 3.26 (m, 4H), 3.83 (m, 2H), 6.17 (d, J=2.1 Hz, 1H), 6.37 (dd, J=3.4, 1.9 Hz, 1H), 6.66 (dd, J=9.1, 2.2 Hz, 1H), 7.01 (m, 2H), 7.31 (m, 2H), 7.48 (m, 3H), 7.78 (dd, J=9.3, 2.3 Hz, 1H), 8.02 (d, J=2.61 Hz, 1H), 8.54 (d, J=2.33 Hz, 1H), 8.58 (t, J=5.9 Hz, 1H, NH), 11.65 (m, 1H).






FINAL COMPD 1
PATENT
| Patent | Submitted | Granted |
|---|---|---|
| APOPTOSIS-INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2014275082] | 2014-02-10 | 2014-09-18 |
| Processes For The Preparation Of An Apoptosis-Inducing Agent [US2014275540] | 2014-03-12 | 2014-09-18 |
| APOPTOSIS INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2010305122] | 2010-12-02 | |
| Panel of micrornas that silence the MCL-1 gene and sensitize cancer cells to ABT-263 [US8742083] | 2010-12-23 | 2014-06-03 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
| METHODS OF TREATMENT USING SELECTIVE BCL-2 INHIBITORS [US2012129853] | 2011-11-22 | 2012-05-24 |
| INHIBITION OF MCL-1 AND/OR BFL-1/A1 [US2015051249] | 2013-03-14 | 2015-02-19 |
| COMBINATION THERAPY OF A TYPE II ANTI-CD20 ANTIBODY WITH A SELECTIVE BCL-2 INHIBITOR [US2014248262] | 2013-09-06 | 2014-09-04 |
References
- New Drugs Online Report for venetoclax
- Hard-to-Treat CLL Yields to Investigational Drug. ASH Dec 2015 refs: Roberts AW, et al "Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia" N Engl J Med 2015; DOI: 10.1056/NEJMoa1513257.
- Phase 2 Study of Venetoclax in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia with 17p Deletion Meets Primary Endpoint
- ABT-199 BH-3 Mimetic Enters Phase Ia Trial For Chronic Lymphocytic Leukemia. 2011
- For Refractory CLL, Venetoclax's Complete Response Rate Is Tops. 2015
External links
- ABT-199 inc formula and structure
References
|
1: Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2
inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 Jan 6. doi: 10.1038/nm.3048. [Epub ahead of print] PubMed PMID: 23291630.
inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 Jan 6. doi: 10.1038/nm.3048. [Epub ahead of print] PubMed PMID: 23291630.
 | |
| Systematic (IUPAC) name | |
|---|---|
| 4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide | |
| Identifiers | |
| CAS Number | 1257044-40-8 |
| PubChem | CID: 49846579 |
| ChemSpider | 29315017 |
| Chemical data | |
| Formula | C45H50ClN7O7S |
| Molecular mass | 868.44 g/mol |
/////////
CC1(CCC(=C(C1)c2ccc(cc2)Cl)CN3CCN(CC3)c4ccc(c(c4)Oc5cc6cc[nH]c6nc5)C(=O)NS(=O)(=O)c7ccc(c(c7)[N+](=O)[O-])NCC8CCOCC8)C
OR
CC1(CCC(=C(C1)C2=CC=C(C=C2)Cl)CN3CCN(CC3)C4=CC(=C(C=C4)C(=O)NS(=O)(=O)C5=CC(=C(C=C5)NCC6CCOCC6)[N+](=O)[O-])OC7=CN=C8C(=C7)C=CN8)C







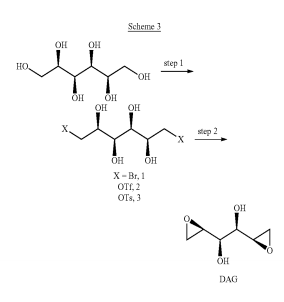
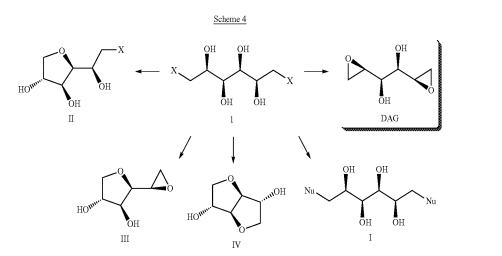
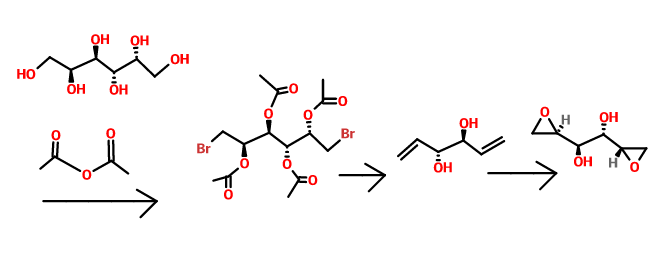
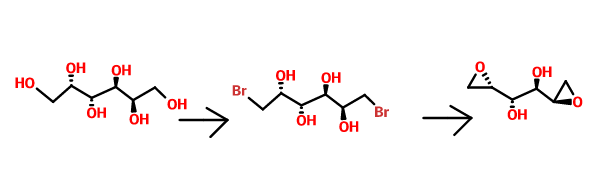 Bromination of dulcitol with HBr at 80°C gives dibromodulcitol , which upon epoxidation in the presence of K2CO3 in t-BuOH or NaOH in H2O or in the presence of ion exchange resin Varion AD (OH) (4) affords the target dianhydrogalactitol .
Bromination of dulcitol with HBr at 80°C gives dibromodulcitol , which upon epoxidation in the presence of K2CO3 in t-BuOH or NaOH in H2O or in the presence of ion exchange resin Varion AD (OH) (4) affords the target dianhydrogalactitol .