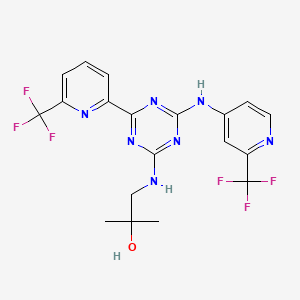
Enasidenib (AG-221)
1446502-11-9
Chemical Formula: C19H17F6N7O
Exact Mass: 473.13988
Chemical Formula: C19H17F6N7O
Exact Mass: 473.13988
AG-221; AG 221; AG221; CC-90007; CC 90007; CC90007; Enasidenib
IUPAC/Chemical Name: 2-methyl-1-((4-(6-(trifluoromethyl)pyridin-2-yl)-6-((2-(trifluoromethyl)pyridin-4-yl)amino)-1,3,5-triazin-2-yl)amino)propan-2-ol
2-methyl-1-(4-(6-(trifluoromethyl)pyridin-2-yl)-6-(2-(trifluoromethyl)pyridin-4-ylamino)-1,3,5-triazin-2-ylamino)propan-2-ol
Agios Pharmaceuticals, Inc. innovator
Enasidenib, aslo known as AG-221 and CC-90007, is a potent and selective IDH2 inhibitor with potential anticancer activity (IDH2 = Isocitrate dehydrogenase 2). The mutations of IDH2 present in certain cancer cells result in a new ability of the enzyme to catalyze the NAPH-dependent reduction of α-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). The production of 2HG is believed to contribute to the formation and progression of cancer . The inhibition of mutant IDH2 and its neoactivity is therefore a potential therapeutic treatment for cancer
AG-221 is an orally available, selective, potent inhibitor of the mutated IDH2 protein, making it a highly targeted investigational medicine for the potential treatment of patients with cancers that harbor an IDH2 mutation. AG-221 has received orphan drug and fast track designations from the U.S. FDA. In September 2013, Agios initiated a Phase 1 multicenter, open-label, dose escalation clinical trial of AG-221 designed to assess the safety and tolerability of AG-221 in advanced hematologic malignancies. In October 2014, Agios initiated four expansion cohorts as part of the ongoing Phase 1 study and expanded its development program with the initiation of a Phase 1/2 study of AG-221 in advanced solid tumors. For the detailed information of AG-221, the solubility of AG-221 in water, the solubility of AG-221 in DMSO, the solubility of AG-221 in PBS buffer, the animal experiment (test) of AG-221, the cell expriment (test) of AG-221, the in vivo, in vitro and clinical trial test of AG-221, the EC50, IC50,and affinity,of AG-221, For the detailed information of AG-221, the solubility of AG-221 in water, the solubility of AG-221 in DMSO, the solubility of AG-221 in PBS buffer, the animal experiment (test) of AG-221, the cell expriment (test) of AG-221, the in vivo, in vitro and clinical trial test of AG-221, the EC50, IC50,and affinity,of AG-221,
Agios Announces New Data from Ongoing Phase 1 Dose Escalation and Expansion Trial of AG-221 Showing Durable Clinical Activity in Patients with Advanced Hematologic Malignancies
IDH2-Mutant Inhibitor Shows Durable Responses of More than 15 Months in Patients with Advanced Acute Myeloid Leukemia (AML) and Other Blood Cancers
Proof-of-Concept Demonstrated in Myelodysplastic Syndrome (MDS) and Untreated AML
125-Patient Expansion Cohort and Global Registration-Enabling Program Remain on Track
Company to Host Conference Call and Webcast Today
CAMBRIDGE, Mass. & VIENNA--(BUSINESS WIRE)--Jun. 12, 2015-- Agios Pharmaceuticals, Inc. (Nasdaq:AGIO), a leader in the fields of cancer metabolism and rare genetic disorders of metabolism, today announced new data from the dose-escalation phase and expansion cohorts from the ongoing Phase 1 study evaluating single agent AG-221, a first-in-class, oral, selective, potent inhibitor of mutant isocitrate dehydrogenase-2 (IDH2), in advanced hematologic malignancies. The data will be presented at the 20th Congress of the European Hematology Association (EHA) taking place June 11-14, 2015 in Vienna.
Data as of May 1, 2015 from 177 patients (104 in dose escalation and 73 from the first four expansion cohorts) with advanced hematologic malignancies treated with single agent AG-221 showed durable clinical activity and a favorable safety profile. More than half of the 177 patients remain on treatment. The study had an overall response rate of 40 percent (63 of 158 response-evaluable patients, using the criteria below) and a complete remission rate of 16 percent (26 of 158 response-evaluable patients). Patients responding to AG-221 continue to show durable clinical activity on treatment for more than 15 months, with an estimated 76 percent of responders staying on treatment for six months or longer. The overall safety profile observed was consistent with previously reported data with more than 100 additional patients treated as of the last analysis.
This new data reflects responses in the evaluable population, which includes all patients with a pre-AG-221 screening assessment and day 28 or later response assessment or an earlier discontinuation for any reason. Patients with a screening assessment who were still on treatment, but had not reached the day 28 disease assessment, were excluded.
“The clinical profile of AG-221 continues to be impressive from the perspectives of response rate, durability, safety and unique mechanism of action,” said Courtney DiNardo, M.D., lead investigator and assistant professor, leukemia atUniversity of Texas MD Anderson Cancer Center. “Additionally, it is encouraging to see early proof-of-concept in myelodysplastic syndrome (MDS) and untreated acute myeloid leukemia (AML) given the need for more effective therapies for these patients.”
“As the data from the AG-221 study continue to mature, we are compiling a robust dataset to quickly move this program into global registration studies later this year in collaboration with Celgene,” said Chris Bowden, M.D., chief medical officer of Agios. “We are excited about the speed of enrollment we’ve seen to date in our four expansion cohorts and are on track to enroll our recently announced fifth expansion cohort of 125 patients with relapsed and/or refractory AML. With this progress, we are executing on our strategy to combine speed and breadth to reach people with hematologic malignancies in urgent need of better treatments.”
About the Ongoing Phase 1 Trial for AG-221 in Advanced Hematologic Malignancies
AG-221 is currently being evaluated in an ongoing Phase 1 trial that includes a dose-escalation phase and four expansion cohorts of 25 patients each, evaluating patients with relapsed or refractory AML who are 60 years of age and older and transplant ineligible; relapsed or refractory AML patients under age 60; untreated AML patients who decline standard of care chemotherapy; and patients with other IDH2-mutant positive hematologic malignancies. Data reported here are from patients receiving AG-221 administered from 60 mg to 450 mg total daily doses in the dose escalation arm and 100 mg once daily in the first four expansion arms, as of May 1, 2015. The median age of these patients is 69 (ranging from 22-90). Treatment with AG-221 showed substantial reduction in the plasma levels of the oncometabolite 2-hydroxglutarate (2HG) to the level observed in healthy volunteers.
Safety Data
A safety analysis was conducted for all 177 treated patients as of May 1, 2015.
- The majority of adverse events reported by investigators were mild to moderate, with the most common being nausea, fatigue, increased blood bilirubin and diarrhea.
- The majority of serious adverse events (SAE) were disease related; SAEs possibly related to study drug were reported in 27 patients.
- A maximum tolerated dose (MTD) has not been reached.
- The all-cause 30-day mortality rate was 4.5 percent.
Efficacy Data
Sixty-three out of 158 response-evaluable patients achieved investigator-assessed objective responses for an overall response rate of 40 percent as of May 1, 2015.
- Of the 63 patients who achieved an objective response, there were 26 (16 percent) complete remissions (CR), three CRs with incomplete platelet recovery (CRp), 14 marrow CRs (mCR), two CRs with incomplete hematologic recovery (CRi) and 18 partial remissions (PR).
- Of the 111 patients with relapsed or refractory AML, 46 (41 percent) achieved an objective response, including 20 (18 percent) CRs, one CRp, 16 PRs, eight mCRs and one CRi.
- Of the 22 patients with AML that had not been treated, seven achieved an objective response, including three CRs, two PRs, one mCR and one CRi.
- Of the 14 patients with myelodysplastic syndrome (MDS), seven achieved an objective response, including two CRs, one CRp and four mCRs.
- Responses were durable, with duration on study drug more than 15 months and ongoing. As of the analysis date, an estimated 88 percent of responses lasted three months or longer, and 76 percent of responses lasted six months or longer.
Upcoming Milestones for AG-221
Agios studies in IDH2-mutated solid and hematologic tumors are ongoing or planned for 2015 to further support development of AG-221.
- Continue to enroll patients in the fifth expansion cohort of 125 patients with IDH2 mutant-positive AML who are in second or later relapse, refractory to second-line induction or re-induction treatment, or have relapsed after allogeneic transplantation.
- Initiate combination trials to evaluate AG-221 as a potential frontline treatment for patients with AML and a broad range of hematologic malignancies in the second half of 2015.
- Initiate a global Phase 3 registration-enabling study in relapsed/refractory AML patients that harbor an IDH2 mutation in the second half of 2015.
- Continue dose escalation in the Phase 1/2 trial in patients with advanced solid tumors, including glioma and angioimmunoblastic T-cell lymphoma (AITL) that carry an IDH2 mutation in 2015.
Conference Call Information
Agios will host a conference call and webcast from the congress to review the data on Friday, June 12, 2015, beginning at 8:00 a.m. ET (2:00 p.m. CEST). To participate in the conference call, please dial (877) 377-7098 (domestic) or (631) 291-4547 (international) and refer to conference ID 53010830. The webcast will be accessible live or in archived form under "Events & Presentations" in the Investors and Media section of the company's website at www.agios.com.
About Agios/Celgene Collaboration
AG-221, the IDH1-mutant inhibitor AG-120 and the pan-IDH mutant inhibitor AG-881 are part of Agios' global strategic collaboration with Celgene Corporation. Under the terms of the collaboration, Celgene has worldwide development and commercialization rights for AG-221. Agios continues to conduct clinical development activities within the AG-221 development program and is eligible to receive up to $120 million in payments on achievement of certain milestones and royalties on net sales. For AG-120, Agios retains U.S. development and commercialization rights. Celgene has an exclusive license outside the United States. Celgene is eligible to receive royalties on net sales in the U.S. Agios is eligible to receive royalties on net sales outside the U.S. and up to $120 million in payments on achievement of certain milestones. For AG-881, the companies have a joint worldwide development and 50/50 profit share collaboration, and Agios is eligible to receive regulatory milestone payments of up to $70 million.
About IDH Mutations and Cancer
IDH1 and IDH2 are two metabolic enzymes that are mutated in a wide range of hematologic and solid tumor malignancies, including AML. Normally, IDH enzymes help to break down nutrients and generate energy for cells. When mutated, IDH increases production of an oncometabolite 2-hydroxyglutarate (2HG) that alters the cells' epigenetic programming, thereby promoting cancer. 2HG has been found to be elevated in several tumor types. Agios believes that inhibition of the mutated IDH proteins may lead to clinical benefit for the subset of cancer patients whose tumors carry them.
About Acute Myelogenous Leukemia (AML)
AML, a cancer of blood and bone marrow characterized by rapid disease progression, is the most common acute leukemia affecting adults. Undifferentiated blast cells proliferate in the bone marrow rather than mature into normal blood cells. AML incidence significantly increases with age, and according to the American Cancer Society, the median age of onset is 66. Less than 10 percent of U.S. AML patients are eligible for bone marrow transplant, and the vast majority of patients do not respond to chemotherapy and progress to relapsed/refractory AML. The five-year survival rate for AML is approximately 20 to 25 percent. IDH2 mutations are present in about 9 to 13 percent of AML cases.
About Myelodysplastic Syndrome (MDS)
MDS comprises a diverse group of bone marrow disorders in which immature blood cells in the bone marrow do not mature or become healthy blood cells. The National Cancer Institute estimates that more than 10,000 people are diagnosed with MDS in the United States each year. Failure of the bone marrow to produce mature healthy cells is a gradual process, and reduced blood cell and/or reduced platelet counts may be accompanied by the loss of the body’s ability to fight infections and control bleeding. For roughly 30 percent of the patients diagnosed with MDS, this bone marrow failure will progress to AML. Chemotherapy and supportive blood products are used to treat MDS.
About Agios Pharmaceuticals, Inc.
Agios Pharmaceuticals is focused on discovering and developing novel investigational medicines to treat cancer and rare genetic disorders of metabolism through scientific leadership in the field of cellular metabolism. In addition to an active research and discovery pipeline across both therapeutic areas, Agios has multiple first-in-class investigational medicines in clinical and/or preclinical development. All Agios programs focus on genetically identified patient populations, leveraging our knowledge of metabolism, biology and genomics. For more information, please visit the company’s website at agios.com.
clips
AG-221, Inhibitor Of IDH2 Mutants

COMBATTING CANCER
Agios’s AG-221 team. Front row (from left): Erin Artin, Kate Yen, Fang Wang, Hua Yang, and Lee Silverman. Back row (from left): Michael Su, Stefan Gross, Sam Agresta, Jeremy Travins, Yue Chen, and Lenny Dang.
Credit: Kevin Graham/Agios
The enzyme isocitrate dehydrogenase (IDH) is probably most famous for its role in the central cellular metabolic pathway, the Krebs cycle. The enzyme catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate. One subtype of the enzyme, IDH1, is found in cells’ cytoplasm, and another, IDH2, is found in their mitochondria.

AG-221
Company: Agios Pharmaceuticals
Target: IDH2
Target: IDH2
People with certain mutations in IDH end up making R-2-hydroxyglutarate (2-HG) instead of α-ketoglutarate. 2-HG is known to make cancer cells flourish. In fact, IDH mutations have been implicated in about 70% of brain cancers and have also been identified in solid tumors and blood cancers, such as acute myeloid leukemia.
Jeremy M. Travins of Agios Pharmaceuticals spoke about how scientists at the company found compounds based on substituted triazines that can cut down on 2-HG production by inhibiting a dimer of mutant IDH2. Using structure-activity relationships and a crystal structure of a lead compound bound to the mutant IDH2 dimer, they managed to develop a clinical candidate: AG-221. It turns out that AG-221 doesn’t bind to the active site of mutant IDH2. Rather, the compound binds to the spot where the two enzymes meet in the dimer.
Hitting this position in just the right way is tricky, Travins explained. Hydrogen-bonding interactions from the triazine and the two amino groups that flank it are critical.
The compound is in Phase I clinical trials, Travins said, and it’s been shown to lower 2-HG levels to those seen in people without cancer. What’s more, he noted, the drug candidate has few side effects, giving patients a higher quality of life than standard chemotherapeutic agents do.
Patent
http://www.google.com/patents/US20130190287
Compound 409—2-methyl-1-(4-(6-(trifluoromethyl)pyridin-2-yl)-6-(2-(trifluoromethyl)pyridin-4-ylamino)-1,3,5-triazin-2-ylamino)propan-2-ol

1H NMR (METHANOL-d4) δ 8.62-8.68 (m, 2H), 847-8.50 (m, 1H), 8.18-8.21 (m, 1H), 7.96-7.98 (m, 1H), 7.82-7.84 (m, 1H), 3.56-3.63 (d, J=28 Hz, 2H), 1.30 (s, 6H). LC-MS: m/z 474.3 (M+H)+.
Patent
http://www.google.com/patents/US20130190287
Compound 409—2-methyl-1-(4-(6-(trifluoromethyl)pyridin-2-yl)-6-(2-(trifluoromethyl)pyridin-4-ylamino)-1,3,5-triazin-2-ylamino)propan-2-ol

1H NMR (METHANOL-d4) δ 8.62-8.68 (m, 2H), 847-8.50 (m, 1H), 8.18-8.21 (m, 1H), 7.96-7.98 (m, 1H), 7.82-7.84 (m, 1H), 3.56-3.63 (d, J=28 Hz, 2H), 1.30 (s, 6H). LC-MS: m/z 474.3 (M+H)+.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2013190287 | 2013-07-25 | THERAPEUTICALLY ACTIVE COMPOUNDS AND THEIR METHODS OF USE |
REFERENCES
1: Caino MC, Altieri DC. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin Cancer Res. 2016 Feb 1;22(3):540-5. doi: 10.1158/1078-0432.CCR-15-0460. Epub 2015 Dec 9. PubMed PMID: 26660517; PubMed Central PMCID: PMC4738153.
2: Stein EM. IDH2 inhibition in AML: Finally progress? Best Pract Res Clin Haematol. 2015 Jun-Sep;28(2-3):112-5. doi: 10.1016/j.beha.2015.10.016. Epub 2015 Oct 19. Review. PubMed PMID: 26590767.
3: Rowe JM. Reasons for optimism in the therapy of acute leukemia. Best Pract Res Clin Haematol. 2015 Jun-Sep;28(2-3):69-72. doi: 10.1016/j.beha.2015.10.002. Epub 2015 Oct 22. Review. PubMed PMID: 26590761.
4: Stein EM. Molecular Pathways: IDH2 Mutations-Co-opting Cellular Metabolism for Malignant Transformation. Clin Cancer Res. 2016 Jan 1;22(1):16-9. doi: 10.1158/1078-0432.CCR-15-0362. Epub 2015 Nov 9. PubMed PMID: 26553750.
5: Kiyoi H. Overview: A New Era of Cancer Genome in Myeloid Malignancies. Oncology. 2015;89 Suppl 1:1-3. doi: 10.1159/000431054. Epub 2015 Nov 10. Review. PubMed PMID: 26551625.
6: Tomita A. [Progress in molecularly targeted therapies for acute myeloid leukemia]. Rinsho Ketsueki. 2015 Feb;56(2):130-8. doi: 10.11406/rinketsu.56.130. Japanese. PubMed PMID: 25765792.
/////////Enasidenib, AG-221,
CC(O)(C)CNC1=NC(C2=NC(C(F)(F)F)=CC=C2)=NC(NC3=CC(C(F)(F)F)=NC=C3)=N1