GSK-2879552
CAS 1401966-69-5 (ABS), 1401966-63-9(REL)
C23 H28 N2 O2, 364.48
Benzoic acid, 4-[[4-[[[(1R,2S)-2-phenylcyclopropyl]amino]methyl]-1-piperidinyl]methyl]-
4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid
- 4-[[4-[[[(1R,2S)-2-Phenylcyclopropyl]amino]methyl]-1-piperidinyl]methyl]benzoic acid
- 4-[[4-[[((1R,2S)-2-Phenylcyclopropyl)amino]methyl]piperidin-1-yl]methyl]benzoic acid
4-((4-((((1R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-1-yl)methyl)benzoic acid
Phase I
Glaxosmithkline Llc INNOVATOR
Neil W. Johnson, Jiri Kasparec, William Henry Miller, Meagan B. Rouse, Dominic Suarez, Xinrong Tian,
A LSD1 inhibitor potentially for the treatment of small cell lung cancer and acute myeloid leukemia.
GSK2879552 is an orally available, irreversible, inhibitor of lysine specific demethylase 1 (LSD1), with potential antineoplastic activity. Upon administration, GSK2879552 binds to and inhibits LSD1, a demethylase that suppresses the expression of target genes by converting the dimethylated form of lysine at position 4 of histone H3 (H3K4) to mono- and unmethylated H3K4. LSD1 inhibition enhances H3K4 methylation and increases the expression of tumor-suppressor genes. This may lead to an inhibition of cell growth in LSD1-overexpressing tumor cells. LSD1, overexpressed in certain tumor cells, plays a key role in tumor cell growth and survival. Check for active clinical trials or closed clinical trials using this agent.

Formula: C23H29ClN2O2
M.Wt: 400.94
M.Wt: 400.94

CAS 1902123-72-1
| Molecular Weight: | 437.41 |
| Formula: | C23H28N2O2.2HCl |
Chromatin modification plays an essential role in transcriptional regulation (T. Kouzarides, 2007, Cell 128: 693-705). These modifications, which include DNA methylation, histone acetylation and hsitone methylation, are disregulated in tumors. This epigenetic disregulation plays an important role in the silencing of tumor suppressors and overexpression of oncogenes in cancer (M. Esteller, 2008, N Engl J Med 358: 1148-59. P. Chi et al, 2010, Nat Rev Cane 10:457-469.). The enzymes that regulate histone methylation are the histone methyl transferases and the histone demethylases.
Lysine-specific demethylase 1 (LSDl; also known as BHC110) is a histone lysine demethylase reported to demethylate H3K4mel/2 (Y. Shi et al, 2004, Cell 119: 941-953) and H3K9mel/2 (R. Schule et al.,2005, Nature 437: 436-439). LSDl is overexpressed in multiple human cancers, including prostate where it is associated with more frequent relapse (P. Kahl et al, 2006, Cane. Res. 66: 11341-11347), breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520) neuroblastoma (J. Kirfel et al, 2009, Cane. Res. 69: 2065-2071. G. Sun et al, 2010, Mol. Cell. Biol. 28: 1997-2000). LSDl is essential for transcriptional regulation mediated by a number of nuclear hormone receptors, including androgen receptor in prostate cancer (R. Schuele et al, 2005, Nature 437: 436-439. R. Schuele et al, 2007, Nat. Cell Biol. 9: 347-353. R. Schuele et al, 2010, Nature 464: 792-796), estrogen receptor in breast carcinomas (M.G. Rosenfeld et al, 2007, Cell 128: 505-518), and TLX receptor in neuorblastoma (S. Kato et al, 2008, Mol. Cell. Biol. 28: 3995-4003). These studies have shown that knockdown of LSDl expression results in decreased cancer cell proliferation. Additionally, LSDl is overexpressed in multiple cancer types that are nuclear hormone receptor-independent. Those tumors include ER-negative breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520), small-cell lung, bladder, head & neck, colon, serous ovary, and kidney Wilm's tumor. Therefore, potent selective small molecule inhibitors of LSDl may be useful for treatment of cancers that are nuclear hormone receptor-dependent and/or nuclear hormone receptor-independent.
The compositions and methods provided herein can potentially be useful for the treatment of cancer including tumors such as skin, breast, brain, cervical carcinomas, testicular carcinomas, etc. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to tumor types such as astrocytic, breast, cervical, colorectal, endometrial, esophageal, gastric, head and neck, hepatocellular, laryngeal, lung, oral, ovarian, prostate and thyroid carcinomas and sarcomas. More specifically, these compounds can potentially be used to treat: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma), small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Kaposi's sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma), large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma); Genitourinary tract: kidney (adenocarcinoma, Wilm's tumor
(nephroblastoma), lymphoma, leukemia), bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma), prostate (adenocarcinoma, sarcoma), testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma); Liver: hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma,angiosarcoma, hepatocellular adenoma, hemangioma; Bone: osteogenic sarcoma(osteosarcoma), fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, malignant giant cell tumor chordoma, osteochronfroma (osteocartilaginous exostoses), benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma and giant cell tumors; Nervous system: skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans), meninges (meningioma, meningiosarcoma, gliomatosis), brain (astrocytoma, meduUoblastoma, glioma, ependymoma, germinoma (pinealoma), glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors), spinal cord neurofibroma, meningioma, glioma, sarcoma); Gynecological: uterus (endometrial carcinoma), cervix (cervical carcinoma, pre -tumor cervical dysplasia), ovaries (ovarian carcinoma (serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma), granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma), vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma), fallopian tubes
(carcinoma); Hematologic: blood (myeloid leukemia (acute and chronic), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplasia syndrome), Hodgkin's disease, non-Hodgkin's lymphoma (malignant lymphoma); Skin: malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Kaposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma. Thus, the term "cancerous cell" as provided herein, includes a cell afflicted by any one of or related to the above identified conditions.
Lysine-specific demethylase 1 (LSDl; also known as BHC110) is a histone lysine demethylase reported to demethylate H3K4mel/2 (Y. Shi et al, 2004, Cell 119: 941-953) and H3K9mel/2 (R. Schule et al.,2005, Nature 437: 436-439). LSDl is overexpressed in multiple human cancers, including prostate where it is associated with more frequent relapse (P. Kahl et al, 2006, Cane. Res. 66: 11341-11347), breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520) neuroblastoma (J. Kirfel et al, 2009, Cane. Res. 69: 2065-2071. G. Sun et al, 2010, Mol. Cell. Biol. 28: 1997-2000). LSDl is essential for transcriptional regulation mediated by a number of nuclear hormone receptors, including androgen receptor in prostate cancer (R. Schuele et al, 2005, Nature 437: 436-439. R. Schuele et al, 2007, Nat. Cell Biol. 9: 347-353. R. Schuele et al, 2010, Nature 464: 792-796), estrogen receptor in breast carcinomas (M.G. Rosenfeld et al, 2007, Cell 128: 505-518), and TLX receptor in neuorblastoma (S. Kato et al, 2008, Mol. Cell. Biol. 28: 3995-4003). These studies have shown that knockdown of LSDl expression results in decreased cancer cell proliferation. Additionally, LSDl is overexpressed in multiple cancer types that are nuclear hormone receptor-independent. Those tumors include ER-negative breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520), small-cell lung, bladder, head & neck, colon, serous ovary, and kidney Wilm's tumor. Therefore, potent selective small molecule inhibitors of LSDl may be useful for treatment of cancers that are nuclear hormone receptor-dependent and/or nuclear hormone receptor-independent.
The compositions and methods provided herein can potentially be useful for the treatment of cancer including tumors such as skin, breast, brain, cervical carcinomas, testicular carcinomas, etc. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to tumor types such as astrocytic, breast, cervical, colorectal, endometrial, esophageal, gastric, head and neck, hepatocellular, laryngeal, lung, oral, ovarian, prostate and thyroid carcinomas and sarcomas. More specifically, these compounds can potentially be used to treat: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma), small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Kaposi's sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma), large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma); Genitourinary tract: kidney (adenocarcinoma, Wilm's tumor
(nephroblastoma), lymphoma, leukemia), bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma), prostate (adenocarcinoma, sarcoma), testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma); Liver: hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma,angiosarcoma, hepatocellular adenoma, hemangioma; Bone: osteogenic sarcoma(osteosarcoma), fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, malignant giant cell tumor chordoma, osteochronfroma (osteocartilaginous exostoses), benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma and giant cell tumors; Nervous system: skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans), meninges (meningioma, meningiosarcoma, gliomatosis), brain (astrocytoma, meduUoblastoma, glioma, ependymoma, germinoma (pinealoma), glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors), spinal cord neurofibroma, meningioma, glioma, sarcoma); Gynecological: uterus (endometrial carcinoma), cervix (cervical carcinoma, pre -tumor cervical dysplasia), ovaries (ovarian carcinoma (serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma), granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma), vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma), fallopian tubes
(carcinoma); Hematologic: blood (myeloid leukemia (acute and chronic), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplasia syndrome), Hodgkin's disease, non-Hodgkin's lymphoma (malignant lymphoma); Skin: malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Kaposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma. Thus, the term "cancerous cell" as provided herein, includes a cell afflicted by any one of or related to the above identified conditions.
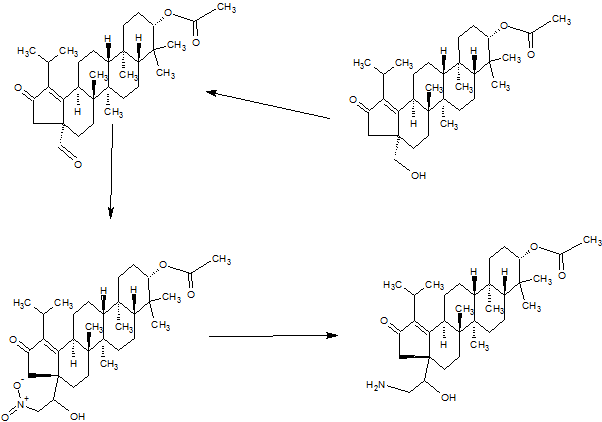
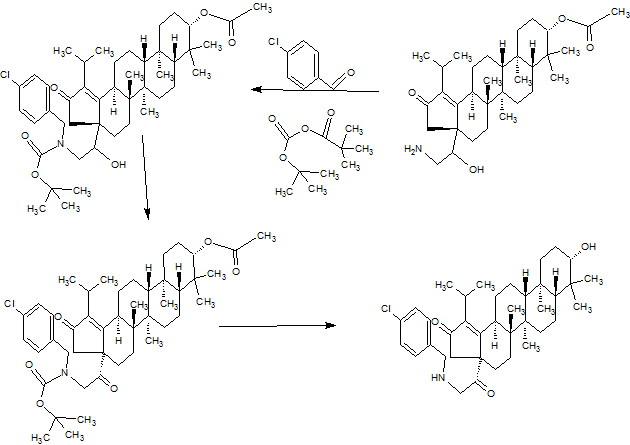
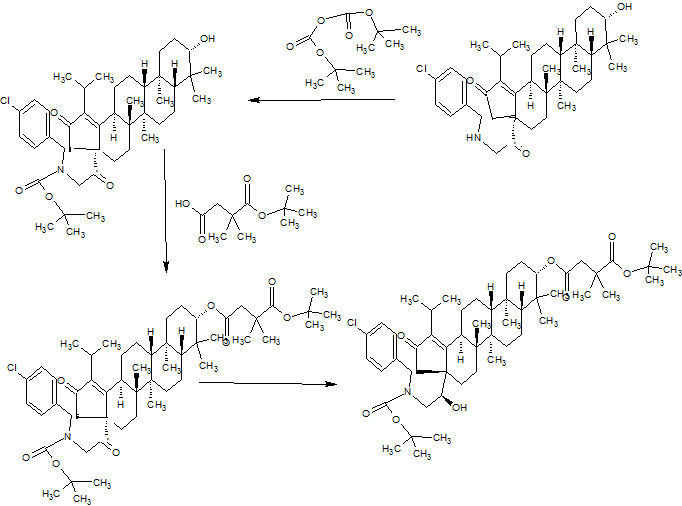
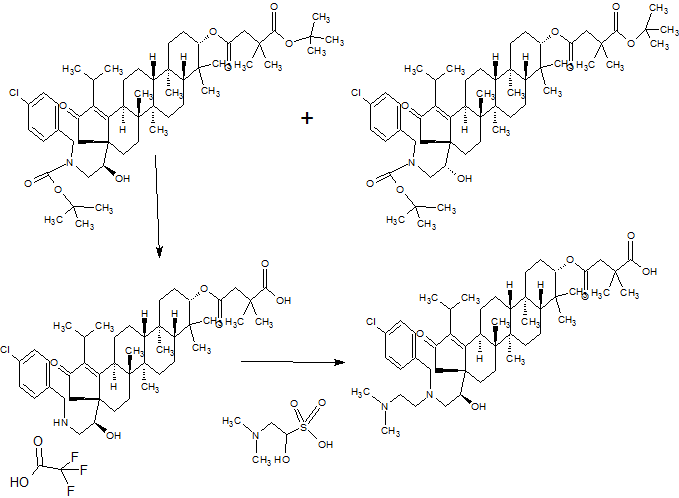
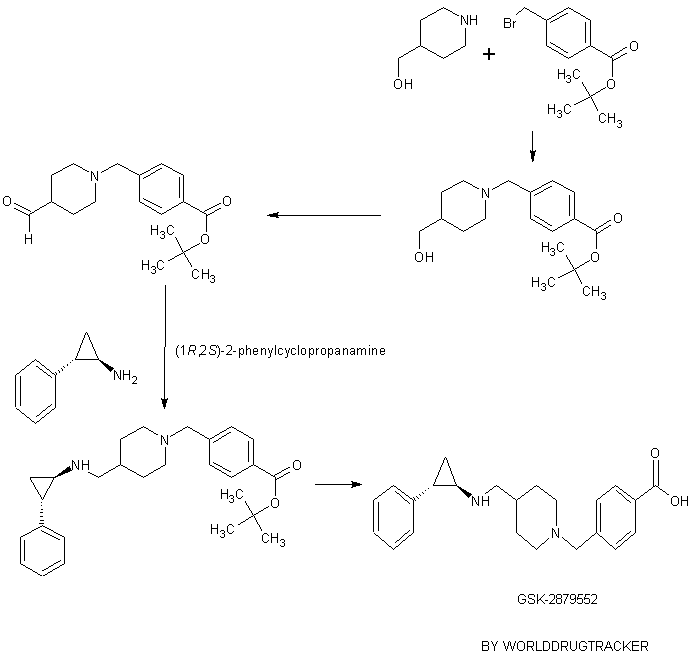
SYNTHESIS
GSK-2879552
PATENT
WO 2012135113
https://www.google.co.in/patents/WO2012135113A2?cl=en
Example 2
1 , 1 -Dimethylethyl 4-( { \( 1 R,2S)-2-phenylcyclopropyl] amino I methyl)- 1 -piperidinecarboxylate

Following a procedure analogous to the procedure described in Example 1 using [(1R,2S)-2-phenylcyclopropyl]amine ((-) isomer) (94 mg, 0.703 mmol) afforded 1,1 -dimethylethyl 4-({[(lR,2S)-2-phenylcyclopropyl]amino}methyl)-l-piperidinecarboxylate (92 mg, 0.264 mmol, 56.4 % yield) as white solid. 1H NMR (400 MHz, METHANOL-d4) δ 7.29 - 7.37 (m, 2H), 7.23 - 7.28 (m, 1H), 7.17 - 7.22 (m, 2H), 4.14 (d, J= 12.63 Hz, 2H), 3.14 (d, J = 7.07 Hz, 2H), 3.01 (dt, J= 4.14, 7.64 Hz, 1H), 2.81 (br. s., 2H), 2.53 (ddd, J= 3.54, 6.63, 10.29 Hz, 1H), 1.97 (ddd, 1H), 1.80 (d, J= 12.13 Hz, 2H), 1.55 (ddd, J= 4.29, 6.63, 10.55 Hz, 1H), 1.47 (s, 9H), 1.36 - 1.45 (m, 1H), 1.23 (qd, J= 4.29, 12.38 Hz, 2H); LC-MS Rt = 0.78 min; MS (ESI): 331.3 [M+H]+.
Example 6
[(lR,2S)-2-Phenylcyclopropyll(4-piperidinylmethyl)amine

Following a procedure analogous to the procedure described in Example 4 using 1,1-dimethylethyl 4-({[(lR,2S)-2-phenylcyclopropyl]amino}methyl)-l-piperidinecarboxylate (Example 2, 60 mg, 0.182 mmol) afforded [(lR,2S)-2-phenylcyclopropyl](4-piperidinylmethyl)amine (41 mg, 0.146 mmol, 80 % yield)as white solid. 1H NMR (400 MHz, METHANOLS) δ 7.29 - 7.38 (m, 2H), 7.23 - 7.29 (m, 1H), 7.18 - 7.23 (m, 2H), 3.47 (d, J= 13.39 Hz, 2H), 3.21 (d, 2H), 2.89 - 3.13 (m, 3H), 2.60 (ddd, J= 3.79, 6.57, 10.36 Hz, 1H), 2.13 - 2.28 (m, J= 3.85, 3.85, 7.61, 11.21 Hz, 1H), 1.99 - 2.13 (m, 2H), 1.49 - 1.71 (m, 3H), 1.35 - 1.48 (m, 1H); LC-MS Rt = 0.44 min; MS (ESI): 231.2
Example 26
4-((4-(((trans-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoic acid

To the solution of 2,2,2-trifluoro-N-(trans-2-phenylcyclopropyl)-N-(piperidin-4-ylmethyl)acetamide (200 mg, 0.613 mmol, Example l ib) and 4-(bromomethyl)benzoic acid (198 mg, 0.919 mmol) in acetonitrile (6 mL) was added potasium carbonate (254 mg, 1.838 mmol). The reaction mixture was stirred for 3 hours at the 90 °C. The reaction mixture was then filtered and evaporated. The crude oil was mixed with 10 mL of 10 % acetic acid and 10 mL of ethyl acetate. Layers were separated, and the organic layer was discharged. Aqueous layer was neutralized with 1 M Na2C03, and the product was extracted into 10 mL of ethyl acetate. The organic layer was washed with brine, dried over MgS04, filtered and evaporated. The oil was dissolved in 6 ml of EtOH and 3 ml of 1 M NaOH. The reaction mixture was stirred for 20 min, and then it was concentrated. The solution was then partioned between 2 ml of water and 5 mL of ethyl acetate. The organic layer was separated and evaporated. The oil was purified on preparatory HPLC (2 to 10 % AcCN: H20 with 0.1 % formic acid modifier). The fractions were collected. To each
fraction was added 1 ml of 1 M HCl, and the fractions were evaporated to dryness. 4-((4-(((trans-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid (50 mg, 0.118 mmol, 19.33 % yield) was isolated as a white solid. 1H NMR (400 MHz,
METHANOLS) δ 8.16 (d, J= 8.34 Hz, 2H), 7.70 (d, J= 8.34 Hz, 2H), 7.30 - 7.37 (m, 2H), 7.23 - 7.29 (m, 1H), 7.20 (d, J= 7.33 Hz, 2H), 4.44 (br. s., 2H), 3.57 (d, J= 11.62 Hz, 2H), 3.07 - 3.27 (m, 4H), 3.04 (dt, J= 3.95, 7.52 Hz, 1H), 2.59 (ddd, J= 3.54, 6.57, 10.11 Hz, lH), 2.12 (d, J= 13.89 Hz, 3H), 1.54 - 1.81 (m, 3H), 1.42 (q, 1H); LC-MS Rt = 0.47 min; MS (ESI): 365.3 [M+H]+.
[M+H]+.
Example 29
4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid

Step 1.
tert-Butyl 4-((4-(hydroxymethyl)piperidin-l-yl)methyl)benzoate
tert-Butyl 4-(bromomethyl)benzoate (1 g, 3.13 mmol) and piperidin-4-ylmethanol (0.361 g, 3.13 mmol) were dissolved in acetonitrile (25 mL). K2CO3 (1.300 g, 9.40 mmol) was added and the reaction mixture was heated to reflux for 20 min. The reaction mixture was cooled down to room temperature, filtered and evaporated. The resulting solid was partitioned between ethyl acetate (50mL) and 1 M HC1 (50 mL). The layers were separated and the aqueous layer was washed with ethyl acetate and the organic layers were discarded. The aqueous layer was basified with 8 M NaOH to pH -10 and extracted 2 times with 50 mL of ethyl acetate. The organic layers were combined, washed with brine and dried over MgSC^, filtered and evaporated. tert-Butyl 4-((4- (hydroxymethyl)piperidin-l-yl)methyl)benzoate (0.95 g, 2.99 mmol, 95 % yield) was isolated as yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.95 (d, J= 8.34 Hz, 2H), 7.39 (d, J = 8.08 Hz, 2H), 3.56 (s, 2H), 3.51 (d, J = 6.57 Hz, 2H), 2.90 (d, J= 11.37 Hz, 2H), 1.94 - 2.04 (m, 2H), 1.73 (d, J= 14.15 Hz, 2H), 1.61 (s, 9H), 1.40 - 1.56 (m, 2H), 1.30 - 1.37 (m, 2H); LC-MS Rt = 0.67 min; MS (ESI): 306.2 [M+H]+.
Step 2.
tert-Butyl 4-((4-formylpiperidin- 1 -yl)methyl)benzoate
To a solution of oxalyl chloride (0.408 mL, 4.67 mmol) in dichloromethane (5 mL) at -60 °C was added a solution of DMSO (0.508 mL, 7.15 mmol) in 15 mL of dichloromethane over 30 minutes. The reaction was stirred for 30 minutes at -60 °C A solution of tert-butyl 4-((4-(hydroxymethyl)piperidin-l-yl)methyl)benzoate (950 mg, 3.11 mmol) in 5 mL of dichloromethane was added over 10 minutes at -60 °C. The reaction mixture was stirred for 3 hours at - 60 °C, then triethylamine (2.168 mL, 15.55 mmol) was added and after 10 minutes 10 mL of water was added. The reaction mixture was allowed to warm up to the room temperature. The layers were separated. The pH of the water layer was adjusted to ~7 with 1 M HC1 and then extracted with 20 mL of dichloromethane. The combined organic layers were washed with water and brine, then dried over MgSO, filtered and evaporated. The resulting oil was purified on a silica column eluting with EtOAc to yield tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (550 mg, 1.722 mmol, 55.4 % yield) as a yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.67 (d, J= 1.26 Hz, 1H), 7.96 (d, J= 8.34 Hz, 2H), 7.38 (d, J= 8.34 Hz, 2H), 3.56 (s, 2H), 2.75 - 2.92 (m, 2H), 2.21 - 2.35 (m, 1H), 2.14 (t, J= 10.48 Hz, 2H), 1.91 (dd, J= 2.78, 13.14 Hz, 2H), 1.65 - 1.81 (m, 2H), 1.58 - 1.64 (m, 9H); LC-MS Rt = 0.69 min; MS (ESI): 304.2
[M+H]+, 322.2 [M+H20]+, 336.6 [M+Na]+
Step 3.
tert-Butyl 4-((4-(((( 1 R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoate
To a solution of tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (6.7 g, 22.08 mmol) in methanol (50 mL) was added (lR,2S)-2-phenylcyclopropanamine (3.53 g, 26.5 mmol). The reaction mixture was refluxed for 5 minutes then cooled down to the room temperature. Sodium cyanotrihydroborate (2.082 g, 33.1 mmol) was added. The reaction mixture was stirred 1 hour at room temperature. Water (50 mL) was added. The reaction was concentrated and 50 mL of dichloromethane was added. The layers were separated. The organics were washed with 10 % acetic acid (50 mL). The layers were separated and 50 mL of brine was added slowly as a solid crashed out. The solid was filtered and suspended in isopropanol. The suspension was sonicated and filtered. tert-Butyl 4-((4-(((( 1 R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoate (5.8 g, 13.65 mmol, 61.8 % yield) was isolated as a white solid. 1H NMR (400 MHz,
METHANOLS) δ 8.07 (d, J= 8.34 Hz, 2H), 7.70 (d, J= 8.08 Hz, 2H), 7.28 - 7.37 (m, 2H), 7.10 - 7.28 (m, 3H), 4.43 (br. s., 2H), 3.54 (d, J= 10.86 Hz, 2H), 3.08 - 3.26 (m, 4H), 3.03 (dt, J= 3.76, 7.39 Hz, 1H), 2.54 - 2.71 (m, 1H), 2.03 - 2.29 (m, 3H), 1.67 - 1.84 (m, 2H), 1.58 - 1.67 (m, 10H), 1.40 (q, J = 6.82 Hz, lH); LC-MS Rt = 0.76 min; MS (ESI): 421.4 [M+H]+.
Step 4.
4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid
A suspension of tert-butyl 4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoate (5.8 g, 13.79 mmol) in HCL - 1 M (80 ml, 80 mmol) was heated to 89 °C (internal temperature) for 2 hr. The solution was cooled down to the room temperature and held in an ice -bath for 1 hour and then filtered. 4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid (3.8 g, 8.25 mmol, 59.8 % yield) was isolated as white solid. 1H NMR (400 MHz, METHANOL-d4) 5 8.15 (d, J= 8.34 Hz, 2H), 7.72 (d, J= 8.59 Hz, 2H), 7.29 - 7.37 (m, 2H), 7.14 - 7.28 (m, 3H), 4.45 (br. s., 2H), 3.55 (d, J= 10.36 Hz, 2H), 3.07 - 3.29 (m, 4H), 3.04 (dt, J= 3.98, 7.71 Hz, 1H), 2.61 (ddd, J= 3.66, 6.57, 10.23 Hz, 1H), 1.98 - 2.31 (m, 3H), 1.72 (br. s., 2H), 1.62 (ddd, J= 4.42, 6.51, 10.55 Hz, 1H), 1.41 (q, J= 6.82 Hz, lH); LC-MS Rt = 0.49 min; MS (ESI): 365.3 [M+H]+.
WO 2012135113
https://www.google.co.in/patents/WO2012135113A2?cl=en
Example 2
1 , 1 -Dimethylethyl 4-( { \( 1 R,2S)-2-phenylcyclopropyl] amino I methyl)- 1 -piperidinecarboxylate
Following a procedure analogous to the procedure described in Example 1 using [(1R,2S)-2-phenylcyclopropyl]amine ((-) isomer) (94 mg, 0.703 mmol) afforded 1,1 -dimethylethyl 4-({[(lR,2S)-2-phenylcyclopropyl]amino}methyl)-l-piperidinecarboxylate (92 mg, 0.264 mmol, 56.4 % yield) as white solid. 1H NMR (400 MHz, METHANOL-d4) δ 7.29 - 7.37 (m, 2H), 7.23 - 7.28 (m, 1H), 7.17 - 7.22 (m, 2H), 4.14 (d, J= 12.63 Hz, 2H), 3.14 (d, J = 7.07 Hz, 2H), 3.01 (dt, J= 4.14, 7.64 Hz, 1H), 2.81 (br. s., 2H), 2.53 (ddd, J= 3.54, 6.63, 10.29 Hz, 1H), 1.97 (ddd, 1H), 1.80 (d, J= 12.13 Hz, 2H), 1.55 (ddd, J= 4.29, 6.63, 10.55 Hz, 1H), 1.47 (s, 9H), 1.36 - 1.45 (m, 1H), 1.23 (qd, J= 4.29, 12.38 Hz, 2H); LC-MS Rt = 0.78 min; MS (ESI): 331.3 [M+H]+.
Example 6
[(lR,2S)-2-Phenylcyclopropyll(4-piperidinylmethyl)amine
Following a procedure analogous to the procedure described in Example 4 using 1,1-dimethylethyl 4-({[(lR,2S)-2-phenylcyclopropyl]amino}methyl)-l-piperidinecarboxylate (Example 2, 60 mg, 0.182 mmol) afforded [(lR,2S)-2-phenylcyclopropyl](4-piperidinylmethyl)amine (41 mg, 0.146 mmol, 80 % yield)as white solid. 1H NMR (400 MHz, METHANOLS) δ 7.29 - 7.38 (m, 2H), 7.23 - 7.29 (m, 1H), 7.18 - 7.23 (m, 2H), 3.47 (d, J= 13.39 Hz, 2H), 3.21 (d, 2H), 2.89 - 3.13 (m, 3H), 2.60 (ddd, J= 3.79, 6.57, 10.36 Hz, 1H), 2.13 - 2.28 (m, J= 3.85, 3.85, 7.61, 11.21 Hz, 1H), 1.99 - 2.13 (m, 2H), 1.49 - 1.71 (m, 3H), 1.35 - 1.48 (m, 1H); LC-MS Rt = 0.44 min; MS (ESI): 231.2
Example 26
4-((4-(((trans-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoic acid
To the solution of 2,2,2-trifluoro-N-(trans-2-phenylcyclopropyl)-N-(piperidin-4-ylmethyl)acetamide (200 mg, 0.613 mmol, Example l ib) and 4-(bromomethyl)benzoic acid (198 mg, 0.919 mmol) in acetonitrile (6 mL) was added potasium carbonate (254 mg, 1.838 mmol). The reaction mixture was stirred for 3 hours at the 90 °C. The reaction mixture was then filtered and evaporated. The crude oil was mixed with 10 mL of 10 % acetic acid and 10 mL of ethyl acetate. Layers were separated, and the organic layer was discharged. Aqueous layer was neutralized with 1 M Na2C03, and the product was extracted into 10 mL of ethyl acetate. The organic layer was washed with brine, dried over MgS04, filtered and evaporated. The oil was dissolved in 6 ml of EtOH and 3 ml of 1 M NaOH. The reaction mixture was stirred for 20 min, and then it was concentrated. The solution was then partioned between 2 ml of water and 5 mL of ethyl acetate. The organic layer was separated and evaporated. The oil was purified on preparatory HPLC (2 to 10 % AcCN: H20 with 0.1 % formic acid modifier). The fractions were collected. To each
fraction was added 1 ml of 1 M HCl, and the fractions were evaporated to dryness. 4-((4-(((trans-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid (50 mg, 0.118 mmol, 19.33 % yield) was isolated as a white solid. 1H NMR (400 MHz,
METHANOLS) δ 8.16 (d, J= 8.34 Hz, 2H), 7.70 (d, J= 8.34 Hz, 2H), 7.30 - 7.37 (m, 2H), 7.23 - 7.29 (m, 1H), 7.20 (d, J= 7.33 Hz, 2H), 4.44 (br. s., 2H), 3.57 (d, J= 11.62 Hz, 2H), 3.07 - 3.27 (m, 4H), 3.04 (dt, J= 3.95, 7.52 Hz, 1H), 2.59 (ddd, J= 3.54, 6.57, 10.11 Hz, lH), 2.12 (d, J= 13.89 Hz, 3H), 1.54 - 1.81 (m, 3H), 1.42 (q, 1H); LC-MS Rt = 0.47 min; MS (ESI): 365.3 [M+H]+.
[M+H]+.
Example 29
4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid
Step 1.
tert-Butyl 4-((4-(hydroxymethyl)piperidin-l-yl)methyl)benzoate
tert-Butyl 4-(bromomethyl)benzoate (1 g, 3.13 mmol) and piperidin-4-ylmethanol (0.361 g, 3.13 mmol) were dissolved in acetonitrile (25 mL). K2CO3 (1.300 g, 9.40 mmol) was added and the reaction mixture was heated to reflux for 20 min. The reaction mixture was cooled down to room temperature, filtered and evaporated. The resulting solid was partitioned between ethyl acetate (50mL) and 1 M HC1 (50 mL). The layers were separated and the aqueous layer was washed with ethyl acetate and the organic layers were discarded. The aqueous layer was basified with 8 M NaOH to pH -10 and extracted 2 times with 50 mL of ethyl acetate. The organic layers were combined, washed with brine and dried over MgSC^, filtered and evaporated. tert-Butyl 4-((4- (hydroxymethyl)piperidin-l-yl)methyl)benzoate (0.95 g, 2.99 mmol, 95 % yield) was isolated as yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.95 (d, J= 8.34 Hz, 2H), 7.39 (d, J = 8.08 Hz, 2H), 3.56 (s, 2H), 3.51 (d, J = 6.57 Hz, 2H), 2.90 (d, J= 11.37 Hz, 2H), 1.94 - 2.04 (m, 2H), 1.73 (d, J= 14.15 Hz, 2H), 1.61 (s, 9H), 1.40 - 1.56 (m, 2H), 1.30 - 1.37 (m, 2H); LC-MS Rt = 0.67 min; MS (ESI): 306.2 [M+H]+.
Step 2.
tert-Butyl 4-((4-formylpiperidin- 1 -yl)methyl)benzoate
To a solution of oxalyl chloride (0.408 mL, 4.67 mmol) in dichloromethane (5 mL) at -60 °C was added a solution of DMSO (0.508 mL, 7.15 mmol) in 15 mL of dichloromethane over 30 minutes. The reaction was stirred for 30 minutes at -60 °C A solution of tert-butyl 4-((4-(hydroxymethyl)piperidin-l-yl)methyl)benzoate (950 mg, 3.11 mmol) in 5 mL of dichloromethane was added over 10 minutes at -60 °C. The reaction mixture was stirred for 3 hours at - 60 °C, then triethylamine (2.168 mL, 15.55 mmol) was added and after 10 minutes 10 mL of water was added. The reaction mixture was allowed to warm up to the room temperature. The layers were separated. The pH of the water layer was adjusted to ~7 with 1 M HC1 and then extracted with 20 mL of dichloromethane. The combined organic layers were washed with water and brine, then dried over MgSO, filtered and evaporated. The resulting oil was purified on a silica column eluting with EtOAc to yield tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (550 mg, 1.722 mmol, 55.4 % yield) as a yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.67 (d, J= 1.26 Hz, 1H), 7.96 (d, J= 8.34 Hz, 2H), 7.38 (d, J= 8.34 Hz, 2H), 3.56 (s, 2H), 2.75 - 2.92 (m, 2H), 2.21 - 2.35 (m, 1H), 2.14 (t, J= 10.48 Hz, 2H), 1.91 (dd, J= 2.78, 13.14 Hz, 2H), 1.65 - 1.81 (m, 2H), 1.58 - 1.64 (m, 9H); LC-MS Rt = 0.69 min; MS (ESI): 304.2
[M+H]+, 322.2 [M+H20]+, 336.6 [M+Na]+
Step 3.
tert-Butyl 4-((4-(((( 1 R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoate
To a solution of tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (6.7 g, 22.08 mmol) in methanol (50 mL) was added (lR,2S)-2-phenylcyclopropanamine (3.53 g, 26.5 mmol). The reaction mixture was refluxed for 5 minutes then cooled down to the room temperature. Sodium cyanotrihydroborate (2.082 g, 33.1 mmol) was added. The reaction mixture was stirred 1 hour at room temperature. Water (50 mL) was added. The reaction was concentrated and 50 mL of dichloromethane was added. The layers were separated. The organics were washed with 10 % acetic acid (50 mL). The layers were separated and 50 mL of brine was added slowly as a solid crashed out. The solid was filtered and suspended in isopropanol. The suspension was sonicated and filtered. tert-Butyl 4-((4-(((( 1 R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoate (5.8 g, 13.65 mmol, 61.8 % yield) was isolated as a white solid. 1H NMR (400 MHz,
METHANOLS) δ 8.07 (d, J= 8.34 Hz, 2H), 7.70 (d, J= 8.08 Hz, 2H), 7.28 - 7.37 (m, 2H), 7.10 - 7.28 (m, 3H), 4.43 (br. s., 2H), 3.54 (d, J= 10.86 Hz, 2H), 3.08 - 3.26 (m, 4H), 3.03 (dt, J= 3.76, 7.39 Hz, 1H), 2.54 - 2.71 (m, 1H), 2.03 - 2.29 (m, 3H), 1.67 - 1.84 (m, 2H), 1.58 - 1.67 (m, 10H), 1.40 (q, J = 6.82 Hz, lH); LC-MS Rt = 0.76 min; MS (ESI): 421.4 [M+H]+.
Step 4.
4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid
A suspension of tert-butyl 4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoate (5.8 g, 13.79 mmol) in HCL - 1 M (80 ml, 80 mmol) was heated to 89 °C (internal temperature) for 2 hr. The solution was cooled down to the room temperature and held in an ice -bath for 1 hour and then filtered. 4-((4-((((lR,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid (3.8 g, 8.25 mmol, 59.8 % yield) was isolated as white solid. 1H NMR (400 MHz, METHANOL-d4) 5 8.15 (d, J= 8.34 Hz, 2H), 7.72 (d, J= 8.59 Hz, 2H), 7.29 - 7.37 (m, 2H), 7.14 - 7.28 (m, 3H), 4.45 (br. s., 2H), 3.55 (d, J= 10.36 Hz, 2H), 3.07 - 3.29 (m, 4H), 3.04 (dt, J= 3.98, 7.71 Hz, 1H), 2.61 (ddd, J= 3.66, 6.57, 10.23 Hz, 1H), 1.98 - 2.31 (m, 3H), 1.72 (br. s., 2H), 1.62 (ddd, J= 4.42, 6.51, 10.55 Hz, 1H), 1.41 (q, J= 6.82 Hz, lH); LC-MS Rt = 0.49 min; MS (ESI): 365.3 [M+H]+.

Neil Johnson
US Lead of Chemistry Talent Development, External Engagement and Recruitment at GSK
Experience
US Lead of Chemistry Talent Development, External Engagement and Recruitment
GSK

Investgator
GlaxoSmithKline

Senior Scientist
Cephalon
Education

The Johns Hopkins University
Doctor of Philosophy (PhD), Organic Chemistry

///////////GSK-2879552, 1401966-63-9, Phase I , A LSD1 inhibitor, small cell lung cancer, acute myeloid leukemia, 1401966-69-5, 1902123-72-1
O=C(O)C1=CC=C(CN2CCC(CN[C@H]3[C@H](C4=CC=CC=C4)C3)CC2)C=C1
O=C(O)c1ccc(cc1)CN2CCC(CC2)CN[C@@H]4C[C@H]4c3ccccc3