Besifloxacin
SS 734, BOL 303224A, ISV-403
MW 430.301, MF C19H21ClFN3O3
141388-76-3 CAS
7-[(3R)-3-aminoazepan-1-yl]-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
(R)-(+)-7-(3-amino-2,3,4,5,6,7-hexahydro-1H-azepin-1-yl)-1,4-dihydro-4-oxoquinoline-3-carboxylic acid
(R) -7- (3- amino-hexahydro-azepin -1H- mushroom-1-yl) -8-chloro-1-cyclopropylmethyl -6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid
Synthesis of the molecule (R)-(+)-7-(3-amino-2,3,4,5,6,7-hexahydro-1H-azepin-1-yl)-1,4-dihydro-4-oxoquinoline-3-carboxylic acid is disclosed in U.S. Pat. No. 5,447,926,
Besifloxacin is a fourth generation fluoroquinolone-type opthalmic antibiotic for the treatment of bacterial conjunctivitis. FDA approved on May 28, 2009. by Bausch & Lomb, for the treatment of non-viral bacterial conjunctivitis
Besifloxacin, (+)-7-[(3R)-3-aminohexahydro-1H-azepin-1-yl]-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride, developed by SS Pharmaceutical (SSP) Co.Ltd. was a fourth-generation fluoroquinolone antibiotic . Besifloxacin hydrochloride eye drop was used to treat bacterial conjunctivitis caused by aerobic and facultative Gram-positive microorganisms and aerobic and facultative Gram-negative microorganisms
Besifloxacin, (+)-7-[(3R)-3-aminohexahydro-1H-azepin-1-yl]-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride, developed by SS Pharmaceutical (SSP) Co.Ltd. was a fourth-generation fluoroquinolone antibiotic . Besifloxacin hydrochloride eye drop was used to treat bacterial conjunctivitis caused by aerobic and facultative Gram-positive microorganisms and aerobic and facultative Gram-negative microorganisms
Besifloxacin (INN/USAN) is a fourth-generation fluoroquinolone antibiotic. The marketed compound is besifloxacin hydrochloride. It was developed by SSP Co. Ltd., Japan, and designated SS734. SSP licensed U.S. and European rights to SS734 for ophthalmic useto InSite Vision Incorporated (OTCBB: INSV) in 2000. InSite Vision developed an eye drop formulation (ISV-403) and conducted preliminary clinical trials before selling the product and all rights to Bausch & Lomb in 2003.[1]
The eye drop was approved by the United States Food and Drug Administration (FDA) on May 29, 2009 and marketed under the trade name Besivance.[2]
| Name | Dosage | Strength | Route | Labeller | Marketing Start | Marketing End | |
|---|---|---|---|---|---|---|---|
| Besivance | suspension | 6 mg/mL | ophthalmic | Bausch & Lomb Incorporated | 2009-05-28 | Not applicable |  |
| Besivance | suspension | 0.6 % | ophthalmic | Bausch & Lomb Inc | 2010-01-27 | Not applicable |  |
| Besivance | suspension | 6 mg/mL | ophthalmic | Physicians Total Care, Inc. | 2011-07-13 | Not applicable | |


405165-61-9 CAS
Besifloxacin Hydrochloride
Besifloxacin hydrochloride is a fourth-generation fluoroquinolone antibiotic.
IC50 Value:
Target: Antibacterial
Besifloxacin has been found to inhibit production of pro-inflammatory cytokines in vitro. Besifloxacin is a novel 8-chloro-fluoroquinolone agent with potent, bactericidal activity against prevalent and drug-resistant pathogens.besifloxacin is the most potent agent tested against gram-positive pathogens and anaerobes and is generally equivalent to comparator fluoroquinolones in activity against most gram-negative pathogens. Besifloxacin demonstrates potent, broad-spectrum activity, which is particularly notable against gram-positive and gram-negative isolates that are resistant to other fluoroquinolones and classes of antibacterial agents.
IC50 Value:
Target: Antibacterial
Besifloxacin has been found to inhibit production of pro-inflammatory cytokines in vitro. Besifloxacin is a novel 8-chloro-fluoroquinolone agent with potent, bactericidal activity against prevalent and drug-resistant pathogens.besifloxacin is the most potent agent tested against gram-positive pathogens and anaerobes and is generally equivalent to comparator fluoroquinolones in activity against most gram-negative pathogens. Besifloxacin demonstrates potent, broad-spectrum activity, which is particularly notable against gram-positive and gram-negative isolates that are resistant to other fluoroquinolones and classes of antibacterial agents.
Clinical Information of Besifloxacin Hydrochloride
| Product Name | Sponsor Only | Condition | Start Date | End Date | Phase | Last Change Date |
|---|---|---|---|---|---|---|
| Besifloxacin Hydrochloride | Bucci Laser Vision Institute | Bacterial infection | 31-MAY-11 | 31-DEC-11 | Phase 4 | 05-JUN-13 |
| Bucci Laser Vision Institute | 31-MAY-11 | 31-DEC-11 | Phase 4 | 03-JUN-13 | ||
| Innovative Medical Services | 30-SEP-10 | 31-OCT-12 | Phase 4 | 11-SEP-13 | ||
| Ophthalmology Consultants, Ltd | Cataract | 30-SEP-10 | 28-FEB-11 | Phase 4 | 11-SEP-13 | |
| University of Louisville | Blepharitis | 31-AUG-11 | 31-OCT-11 | Phase 4 | 01-DEC-11 |
Pharmacodynamics
Besifloxacin is a fluoroquinolone that has a broad spectrum in vitro activity against a wide range of Gram-positive and Gram-negativeocular pathogens: e.g., Corynebacterium pseudodiphtheriticum, Moraxella lacunata, Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus hominis, Streptococcus mitis, Streptococcus oralis, Streptococcus pneumoniae and Streptococcus salivarius. Besifloxacin has been found to inhibit production of pro-inflammatory cytokines in vitro.[3] The mechanism of action of besifloxacin involves inhibition of two enzymes which are essential for the synthesis and replication of bacterial DNA: the bacterialDNA gyrase and topoisomerase IV.
Medical Use
Besifloxacin is indicated in the treatment of bacterial conjunctivitis caused by sensitive germs,[4] as well as in the prevention of infectious complications in patients undergoing laser therapy for the treatment of cataracts.[5][6]
Adverse Effects
During the treatment, the most frequently reported ocular adverse reaction was the appearance of conjunctival redness (approximately 2% of patients). Other possible adverse reactions, reported in subjects treated with besifloxacin were: eye pain, itching of the eye, blurred vision, swelling of the eye or eyelid.
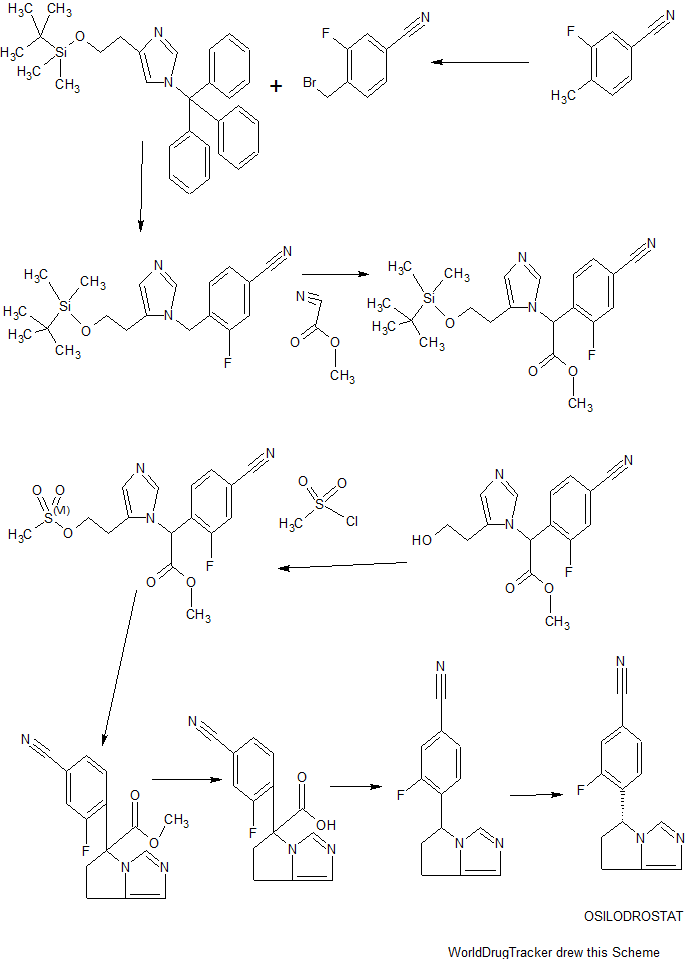
MORE SYNTHESIS COMING, WATCH THIS SPACE…………………..

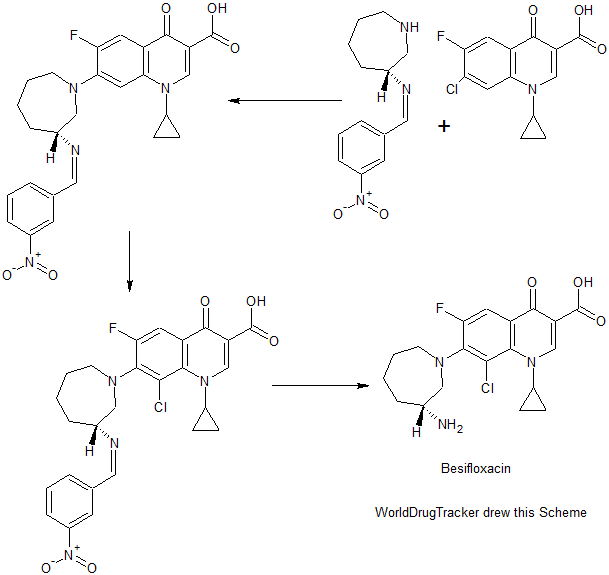
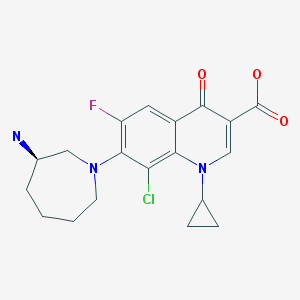
PATENT
WO 2010111116
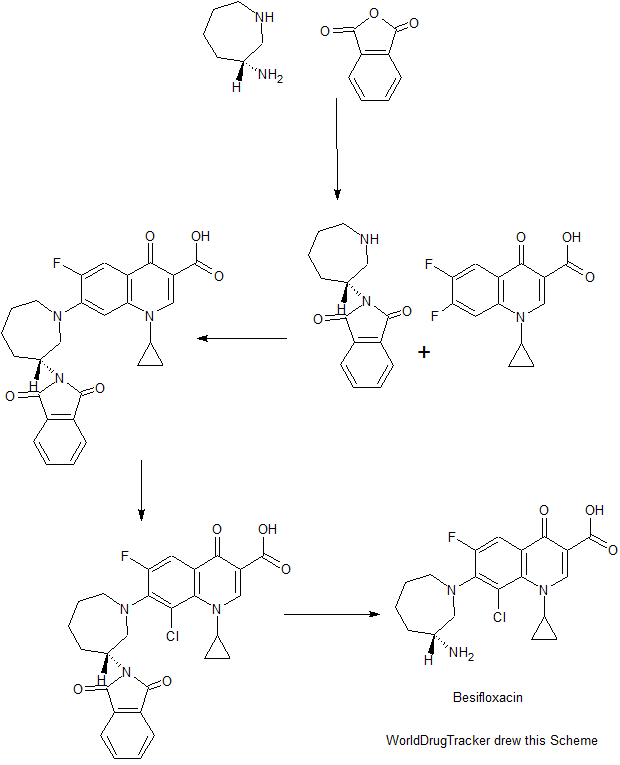
PATENT
CN 104592196
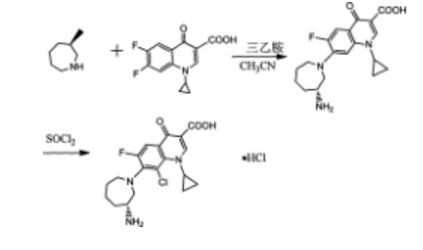
The method comprises performing condensation reaction of 1-cyclopropyl-6,7-dichloro-1,4-dihydro-4-oxy-3-quinoline carboxylic acid with (R)-3-aminohexahydroazepine in the presence of org. base in org. solvent I at 45°C-solvent b.p. temp. under refluxing, washing with acid, vacuum concg. to obtain (R)-7-(3-amino-hexahydro-1H-azepine-1-yl)-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxy-3-quinoline carboxylic acid, dissolving in 5-10 fold org. solvent II, reacting with thionyl chloride at 0-40°C, and vacuum concg. to obtain (R) -7- (3- amino-hexahydro-azepin -1H- mushroom-1-yl) -8-chloro-1-cyclopropylmethyl -6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
Preparation method of the present invention provides hydrochloride Besifloxacin, comprising the steps of:
(1), in three _6 flask of 1-cyclopropyl, 6,7-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid 10g of acetonitrile added 100mL, was added (R ) -3-amino-hexahydro-aza mushroom 4.73g and 7.2mL of triethylamine was heated at reflux for 5h TLC plate detection point, the reaction was complete spin dry plus 100mL dissolved in chloroform and then 200mL 1M hydrochloric acid and washed twice with saturated brine The organic phase to pH 4-6, the organic phase was poured into the jar and dried to obtain the single (R) -7- (3- amino-hexahydro-azepin -1H- leather-yl) cyclopropyl-6 -1_ fluoro-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid in chloroform solution; spin-dried to give (R) -7- (3- amino-hexahydro-azepin -1H- leather-yl) cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-quinoline-3-carboxylic acid.
(2), obtained in the previous step (R) -7- (3_ atmosphere atmosphere -1H- gas hybrid group six leather-1-yl) cyclopropyl-6-fluoro-1,4 _1_ dihydro-4-oxo-3-quinolinecarboxylic acid in chloroform solution was cooled to 0 ° C, was slowly added dropwise under constant stirring 18mL S0C12, temperature does not exceed 5 ° C added, the mixture was stirred at 0 ° C after 2h l to room temperature, TLC detection, after completion of the reaction was evaporated to dryness to column chromatography to give (R) -7- (3- amino-hexahydro-azepin -1H- mushroom-1-yl) -8-chloro-1-cyclopropylmethyl -6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride 5. 12g.
PATENT
US 20110144329
EXAMPLE 1Preparation of Besifloxacin Free Base Solid
Besifloxacin free base was prepared from besifloxacin hydrochloride addition salt.
An amount of about 5 g of besifloxacin HCl (HCl addition salt of besifloxacin made, for example, by the method of U.S. Pat. No. 5,447,926; which is incorporated herein by reference in its entirety) was added to about 750 ml of water. The besifloxacin HCl was allowed to dissolve in said water. Twenty milliliters of 1N NaOH solution were added slowly to the besifloxacin aqueous solution while stirring (final pH 10.2). Besifloxacin free base started to precipitate. Eight milliliters of 1N HCl solution were added slowly while stirring (final pH of 9.7). The resulting mixture was allowed to mix for 2 hours while besifloxacin free base continued to precipitate. At the end of 2 hours, the precipitated besifloxacin free base was filtered through a Millipore type RA 1.2 μm filter. The besifloxacin free base thus collected was dried in a vacuum oven at room temperature. 4.35 g of besifloxacin free base was recovered.
FIG. 1 shows a UV absorption spectrum of besifloxacin free base starting material of Example 1.

FIG. 3 shows an IR spectrum of free base starting material of Example 1.

PATENT


Example 6 (R) -7_ (3- amino-hexahydro--1H- diazepan-1-yl) -8_ chloro-1-cyclopropyl-6-fluoro-1,4- Hydrogen oxo - quinoline-3-carboxylic acid (Besifloxacin). [0021] The reaction vessel was added chloroform (50ml) as a reaction solvent, in the case of a solid material was added with stirring (III) (3. 59g, O. Olmol), until the intermediate (III) is completely dissolved, was added dropwise under ice- chlorosulfonic acid, stirred for I hour under ice-cooling, gradually warmed to room temperature, stirred for 6 hours, and then reacted at reflux temperature for 6 hours. After completion of the reaction by TLC, the reaction solution was cooled to 0 ° C, white solid was precipitated, filtered, washed with a small amount of dichloromethane to give a crude product besifloxacin (3. 65g, 93. 01%). [0022] Example 7 (R) -7_ (3- amino-hexahydro--1H- diazepan-1-yl) -8_ chloro-1-cyclopropyl-6-fluoro-1,4- Hydrogen oxo - quinoline-3-carboxylic acid (Besifloxacin). [0023] The reaction vessel was added chloroform (50ml) as a reaction solvent, in the case of a solid material was added with stirring (III) (3. 59g, 0. Olmol), until the intermediate (III) is completely dissolved, was added dropwise under ice- chlorosulfonic acid was stirred for I hour under ice-cooling, gradually warmed to room temperature, stirred for 6 hours, and then reacted at reflux temperature for 12 hours. After completion of the reaction by TLC, the reaction solution was cooled to 0 ° C, the precipitated white solid was filtered , washed with a little dichloromethane to give Besifloxacin crude (3. 05g, 77. 22%).
PAPER
Molbank 2013, 2013(2), M801; doi:10.3390/M801
Short Note
(R)-7-(Azepan-3-ylamino)-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid Hydrochloride
Supplementary File 3:Support Information (PDF, 340 KB)
Download PDF [188 KB, 27 May 2013; original version 22 May 2013]
R&D Center, Jiangsu Yabang Pharmaceutical Group, Changzhou 213200, China
In this paper (R)-7-(azepan-3-ylamino)-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride 1was isolated and identified as the N-substituted regioisomer of besifloxacin, which has been synthesized from the reaction of 8-chloro-1-cyclopropyl-6,7-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 3 with (R)-tert-butyl 3-aminoazepane-1-carboxylate 2in acetonitrile as solvent in 37% yield. The chemical structure of compound 1 was established on the basis of 1H-NMR, 13C-NMR, mass spectrometry data and elemental analysis
 REGIOMER OF BESIFLOXACINBESIFLOXACIN
REGIOMER OF BESIFLOXACINBESIFLOXACIN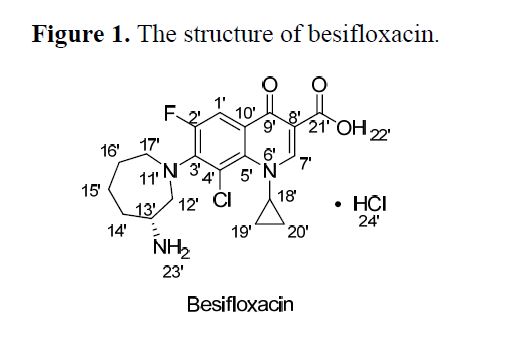
References
- "InSite Vision Reaches Agreement to Sell ISV-403 to Bausch & Lomb" (Press release). InSite Vision. 2003-12-19. Retrieved 2009-08-15.
- "Bausch & Lomb Receives FDA Approval of Besivance, New Topical Ophthalmic Antibacterial for the Treatment of Bacterial Conjunctivitis ("Pink Eye")" (Press release). Bausch & Lomb. 2009-05-29. Retrieved 2009-05-29.
- Zhang JZ, Ward KW (January 2008). "Besifloxacin, a novel fluoroquinolone antimicrobial agent, exhibits potent inhibition of pro-inflammatory cytokines in human THP-1 monocytes". J. Antimicrob. Chemother. 61 (1): 111–6. doi:10.1093/jac/dkm398. PMID 17965029.
- Malhotra R, Ackerman S, Gearinger LS, Morris TW, Allaire C (December 2013). "The safety of besifloxacin ophthalmic suspension 0.6 % used three times daily for 7 days in the treatment of bacterial conjunctivitis". Drugs in R&D 13 (4): 243–52. doi:10.1007/s40268-013-0029-1. PMC 3851703. PMID 24142473. Retrieved 2015-01-06.
- Majmudar PA, Clinch TE (May 2014). "Safety of besifloxacin ophthalmic suspension 0.6% in cataract and LASIK surgery patients". Cornea33 (5): 457–62. doi:10.1097/ICO.0000000000000098. PMC 4195578. PMID 24637269. Retrieved 2015-01-06.
- Nielsen SA, McDonald MB, Majmudar PA (2013). "Safety of besifloxacin ophthalmic suspension 0.6% in refractive surgery: a retrospective chart review of post-LASIK patients". Clinical Ophthalmology (Auckland, N.Z.) 7: 149–56. doi:10.2147/OPTH.S38279. PMC 3552478. PMID 23355771. Retrieved 2015-01-06.
CLIPS
Besifloxacin hydrochloride (Besivance) Besifloxacin is a fourth-generation fluoroquinolone antibiotic which is marketed as besifloxacin hydrochloride. It was originally developed by the Japanese firm SSP Co. Ltd and designated SS734. SSP then licensed U.S. and European rights of SS734 for ophthalmic use to InSite Vision, Inc., in 2000, who then developed an eye drop formulation (ISV-403) and conducted preliminary clinical trials before selling the product and all rights to Bausch & Lomb in 2003.
The eye drop was approved by the United States Food and Drug Administration (FDA) on May 29, 2009 and marketed under the trade name Besivance.24a
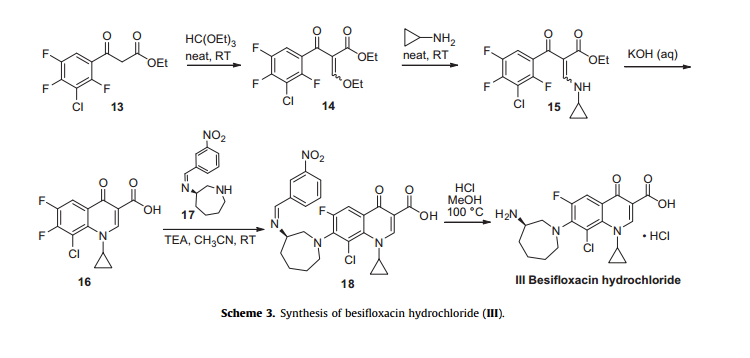
Besifloxacin has been found to inhibit production of pro-inflammatory cytokines in vitro. The synthesis of besifloxacin commences with commercially available ethyl 3-(3-chloro-2,4,5-trifluorophenyl)-3-oxopropanoate (13, Scheme3).24b
Condensation of this ketoester with triethyl orthoformate resulted in a mixture of vinylogous esters 14. Substitution with cyclopropanamine converts 14 to the vinylogous amide 15 as an unreported distribution of cis- and trans-isomers. This mixture was treated with base at elevated temperature to give 16.
Presumably, the trans-isomer isomerizes to the cis-isomer, which subsequently undergoes an intramolecular nucleophilic aromatic substitution with concomitant saponification to construct quinolone acid 16.
Quinolone 16 is then subjected to another nucleophilic substitution involving readily available iminoazepine 17 and the displacement reaction proceeds regioselectively to furnish the atomic framework of besifloxacin (18).
Acidic methanolysis of 18 at elevated temperature gave besiflozacin (III).
24. (a) Bertino, J. S.; Zhang, J.-Z. Expert Opin. Pharmacother. 2009, 10, 2545; (b) Harms, A. E.; Arul, R.; Soni, A. K. U.S. 2009561283 A1, 2009.
| US5447926 * | Sep 16, 1994 | Sep 5, 1995 | Ss Pharmaceutical Co., Ltd. | Quinolone carboxylic acid derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN104458945A * | Nov 27, 2014 | Mar 25, 2015 | 广东东阳光药业有限公司 | Separation and measurement method of besifloxacin hydrochloride and isomer of besifloxacin hydrochloride |
| CN102659761A * | Apr 27, 2012 | Sep 12, 2012 | 常州亚邦制药有限公司 | Method for preparing besifloxacin hydrochloride |
| US5385900 * | Nov 8, 1993 | Jan 31, 1995 | Ss Pharmaceutical Co., Ltd. | Quinoline carboxylic acid derivatives |
| Reference | ||
|---|---|---|
| 1 | * | 黄山等: "克林沙星的 2, 4, 5-三氟苯甲酸路线合成", 《中国医药工业杂志》, vol. 31, no. 8, 31 December 2000 (2000-12-31) |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN103709100A * | Dec 31, 2013 | Apr 9, 2014 | 南京工业大学 | Preparation method of 8-chloroquinolone derivatives |
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
| 7-[(3R)-3-Aminoazepam-1-yl]-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid | |
| Clinical data | |
| Trade names | Besivance |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a610011 |
| License data |
|
| Routes of administration | Ophthalmic |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 141388-76-3 |
| ATC code | S01AE08 (WHO) |
| PubChem | CID 10178705 |
| ChemSpider | 8354210 |
| UNII | BFE2NBZ7NX |
| ChEMBL | CHEMBL1201760 |
| Chemical data | |
| Formula | C19H21ClFN3O3 |
| Molar mass | 393.84 g·mol−1 |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US5,447,926 | No | 1995-09-05 | 2012-09-05 | |
| US5447926 | No | 1996-04-13 | 2016-04-13 | |
| US6,685,958 | No | 2004-02-03 | 2021-06-20 | |
| US6,699,492 | No | 2004-03-02 | 2019-03-31 | |
| US6685958 | No | 2001-06-29 | 2021-06-29 | |
| US6699492 | No | 1999-03-31 | 2019-03-31 | |
| US8415342 | No | 2010-11-07 | 2030-11-07 | |
| US8481526 | No | 2011-01-09 | 2031-01-09 | |
| US8604020 | No | 2010-03-12 | 2030-03-12 | |
| US8937062 | No | 2009-11-13 | 2029-11-13 | |
- O'Brien TP: Besifloxacin ophthalmic suspension, 0.6%: a novel topical fluoroquinolone for bacterial conjunctivitis. Adv Ther. 2012 Jun;29(6):473-90. doi: 10.1007/s12325-012-0027-7. Epub 2012 Jun 20. [PubMed:22729919 ]
- Proksch JW, Granvil CP, Siou-Mermet R, Comstock TL, Paterno MR, Ward KW: Ocular pharmacokinetics of besifloxacin following topical administration to rabbits, monkeys, and humans. J Ocul Pharmacol Ther. 2009 Aug;25(4):335-44. doi: 10.1089/jop.2008.0116. [PubMed:19492955 ]
Besifloxacin Hydrochloride
[1]. Wang Z, Wang S, Zhu F, Chen Z, Yu L, Zeng S. Determination of enantiomeric impurity in besifloxacin hydrochloride by chiral high-performance liquid chromatography with precolumn derivatization. Chirality. 2012 Jul;24(7):526-31. doi: 10.1002/chir.22042.
Abstract
Besifloxacin hydrochloride is a novel chiral broad-spectrum fluoroquinolone developed for the treatment of bacterial conjunctivitis. R-besifloxacin hydrochloride is used in clinics as a consequence of its higher antibacterial activity. To establish an enantiomeric impurity determination method, some chiral stationary phases (CSPs) were screened. Besifloxacin enantiomers can be separated to a certain extent on Chiral CD-Ph (Shiseido Co., Ltd., Japan), Chiral AGP, and Crownpak CR (+) (Daicel Chemical IND., Ltd., Japan). However, the selectivity and sensitivity were both unsatisfactory on these three CSPs. Therefore, Chiral AGP, Chiral CD-Ph, and Crownpak CR (+) were not used in the enantiomeric impurity determination of besifloxacin hydrochloride. The separation of enantiomers of besifloxacin was further performed using a precolumn derivatization chiral high-performance liquid chromatography method. 2,3,4,6-Tetra-O-acetyl-beta-D-glucopyranosyl isothiocyanate was used as the derivatization reagent. Besifloxacin enantiomer derivates were well separated on a C(18) column (250 × 4.6 mm, 5 μm) with a mobile phase that consisted of methanol-KH(2)PO(4) buffer solution (20 mM; pH 3.0) (50:50, v/v). Selectivity, sensitivity, linearity, accuracy, precision, stability, and robustness of this method were all satisfied with the method validation requirement. The method was suitable for the quality control of enantiomeric impurity in besifloxacin hydrochloride.
[2]. Hussar DA. New drugs: golimumab, besifloxacin hydrochloride, and artemether/lumefantrine. J Am Pharm Assoc (2003). 2009 Jul-Aug;49(4):570-4.
[3]. Nafziger AN, Bertino JS Jr. Besifloxacin ophthalmic suspension for bacterial conjunctivitis. Drugs Today (Barc). 2009 Aug;45(8):577-88.
Abstract
Besifloxacin hydrochloride ophthalmic suspension 0.6% (Besivance) is a recently approved fluoroquinolone for the topical treatment of bacterial conjunctivitis. The drug is rapidly bactericidal against common bacterial pathogens causing conjunctivitis, i.e., coagulase-negative Staphylococcus, Streptococcus pneumoniae, Staphylococcus aureus and Haemophilus influenzae as well as against other less common organisms. In addition to being a potent agent against Gram-positive and Gram-negative pathogens including those resistant to other fluoroquinolones, besifloxacin has balanced DNA gyrase and topoisomerase IV activity, which should slow the development of resistance. Topical administration achieves high sustained concentrations in human tears and good ocular tissue penetration in animals while demonstrating an excellent safety profile. Besifloxacin's pharmacokinetic and pharmacodynamic characteristics meet the criteria for successful eradication of many Gram-positive and Gram-negative bacteria while demonstrating minimal systemic exposure. The biochemical properties, achievement of target pharmacokinetic/pharmacodynamic goals and the restriction of besifloxacin to topical ophthalmic use should result in slower development of bacterial resistance, making besifloxacin a new, appealing option for empiric therapy in acute bacterial conjunctivitis.
[4]. Proksch JW, Ward KW. Ocular pharmacokinetics/pharmacodynamics of besifloxacin, moxifloxacin, and gatifloxacin following topical administration to pigmented rabbits. J Ocul Pharmacol Ther. 2010 Oct;26(5):449-58.
Abstract
PURPOSE: The purpose of this investigation was to evaluate the ocular pharmacokinetic/pharmacodynamic (PK/PD) relationship for besifloxacin, moxifloxacin, and gatifloxacin using rabbit ocular PK data, along with in vitro minimum inhibitory concentration (MIC90) values against methicillin- and ciprofloxacin-resistant Staphylococcus aureus (MRSA-CR) and Staphylococcus epidermidis (MRSE-CR).METHODS: Rabbits received a topical instillation of Besivance? (besifloxacin ophthalmic suspension, 0.6%), Vigamox (moxifloxacin hydrochloride ophthalmic solution, 0.5% as base), or Zymar (gatifloxacin ophthalmic solution, 0.3%), and ocular tissues and plasma were collected from 4 animals/treatment/collection time at 8 predetermined time intervals during the 24h after dosing. Ocular levels of each agent were measured by LC/MS/MS, and PK parameters (Cmax, Tmax, and AUC????) were determined. AUC????/MIC?? ratios were calculated for tears, conjunctiva, cornea, and aqueous humor using previously reported MIC??values for MRSA-CR and MRSE-CR.RESULTS: All of the fluoroquinolones tested demonstrated rapid penetration into ocular tissues after a single instillation. Besifloxacin demonstrated the highest exposure in tear fluid, while exposure in conjunctiva was comparable for all 3 compounds. Peak concentrations of all fluoroquinolones in aqueous humor were at or below ~1g/mL. In comparison with their MIC??values against MRSE-CR and MRSA-CR, besifloxacin achieved an AUC????/MIC?? ratio of ~800 in tears, compared with values of ≤10 for moxifloxacin and gatifloxacin. In cornea, conjunctiva, and aqueous humor, the AUC????/MIC?? ratios were <10 for all compounds. However, in these tissues AUC????/MIC?? ratios for besifloxacin were 1.5- to 38-fold higher than moxifloxacin and gatifloxacin....
[5]. Comstock TL, Paterno MR, Usner DW, Pichichero ME. Efficacy and safety of besifloxacin ophthalmic suspension 0.6% in children and adolescents with bacterial conjunctivitis: a post hoc, subgroup analysis of three randomized, double-masked, parallel-group, multicenter clinical trials. Paediatr Drugs. 2010 Apr 1;12(2):105-12. doi: 10.2165/11534380-000000000-00000.
Abstract
BACKGROUND: Acute conjunctivitis is the most frequent eye disorder seen by primary care physicians and one that often affects children. Besifloxacin is a new topical fluoroquinolone, the first chlorofluoroquinolone, for the treatment of bacterial conjunctivitis.OBJECTIVE: To examine the efficacy and safety of besifloxacin ophthalmic suspension 0.6% in patients aged 1-17 years with bacterial conjunctivitis.METHODS: This was a post hoc analysis of a subgroup of pediatric patients aged 1-17 years who had participated in three previously reported, randomized, double-masked, parallel-group, multicenter, clinical trials evaluating the safety and efficacy of besifloxacin in the treatment of bacterial conjunctivitis. The studies were conducted in a community setting (clinical centers). All three clinical trials included children (aged > or = 1 year) with a clinical diagnosis of bacterial conjunctivitis in at least one eye, based on the presence at baseline of grade 1 or greater purulent conjunctival discharge and conjunctival injection, and pin-hole visual acuity of at least 20/200 in both eyes for verbal patients. Two trials were vehicle controlled; the third trial was comparator controlled (moxifloxacin hydrochloride ophthalmic solution 0.5% as base). In all studies, besifloxacin ophthalmic suspension 0.6% was administered as one drop in the affected eye(s) three times daily, at approximately 6-hourly intervals, for 5 days. The main outcome measures were clinical resolution and microbial eradication at visit 2 (day 4 +/- 1 in one study; day 5 +/- 1 in the other two studies) and visit 3 (day 8 or 9). Data from the two vehicle-controlled studies were combined for the assessments to provide greater statistical power.RESULTS: This analysis included 815 pediatric patients aged 1-17 years (447 with culture-confirmed bacterial conjunctivitis). Clinical resolution was significantly greater (p < 0.05) in the besifloxacin group than in the vehicle group at both visit 2 (53.7% vs 41.3%) and visit 3 (88.1% vs 73.0%). Similarly, microbial eradication was significantly higher with besifloxacin than with vehicle at visit 2 (85.8% vs 56.3%) and visit 3 (82.8% vs 68.3%). No significant differences in clinical resolution and microbial eradication were noted between besifloxacin and moxifloxacin. Besifloxacin was well tolerated, with similar incidences of adverse events in the besifloxacin, vehicle, and moxifloxacin groups.CONCLUSION: Besifloxacin ophthalmic suspension 0.6% was shown to be safe and effective for the treatment of bacterial conjunctivitis in children and adolescents aged 1-17 years.
///////Besifloxacin hydrochloride, Besivance, Besifloxacin, SS734, 141388-76-3, 405165-61-9, BOL 303224A, ISV-403, Bausch & Lomb, treatment of non-viral bacterial conjunctivitis
Fc1c(c(Cl)c2c(c1)C(=O)C(\C(=O)O)=C/N2C3CC3)N4CCCC[C@@H](N)C4