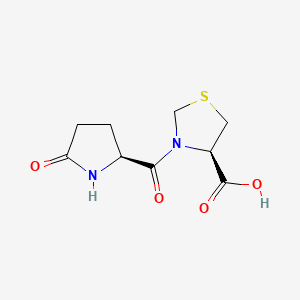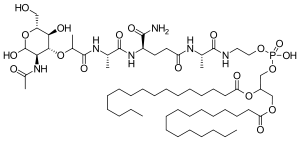
Mifamurtide (Mepact )
- MF C59H109N6O19P
- MW 1237.499
CGP-19835, MFCD09954133, MTP-cephalin, Mtp-PE
Muramyl tripeptide phosphatidylethanolamine
N-(N-Acetylmuramoyl)-L-alanyl-D-α-glutaminyl-N-[(7R)-4-hydroxy-4-oxido-10-oxo-7-[(1-oxohexadecyl)oxy]-3,5,9-trioxa-4-phosphapentacos-1-yl]-L-alaninamide
N-Acetylmuramyl-L-alanyl-D-isoglutamine-L-alanine 2-(1',2'-dipalmitoyl-sn-glycero-3'-hydroxyphosphoryloxy)ethylamide
(2R,5S,8R,13S,22R)-2-{[(3R,4R,5S,6R)-3-Acetamido-2,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-4-yl]oxy}-8-carbamoyl-19-hydroxy-5,13-dimethyl-19-oxido-3,6,11,14,25-pentaoxo-18,20,24-trioxa-4,7,12 ;,15-tetraaza-19λ5-phosphatetracontan-22-yl hexadecanoate
Mifamurtide (trade name Mepact, marketed by Takeda) is a drug against osteosarcoma, a kind of bone cancer mainly affecting children and young adults, which is lethal in about a third of cases. The drug was approved in Europe in March 2009.

History
The drug was invented by Ciba-Geigy (now Novartis) in the early 1980s and sold to Jenner Biotherapies in the 1990s. In 2003,IDM Pharma bought the rights and developed it further.[1] IDM Pharma was acquired by Takeda along with mifamurtide in June 2009.[2]
Mifamurtide had already been granted orphan drug status by the U.S. Food and Drug Administration (FDA) in 2001, and theEuropean Medicines Agency (EMA) followed in 2004. It was approved in the 27 European Union member states plus Iceland, Liechtenstein, and Norway by a centralized marketing authorization in March 2009. The drug was denied approval by the FDA in 2007.[3][4] Mifamurtide has been licensed by the EMA since March, 2009.[5]
Indications
Mifamurtide is indicated for the treatment of high-grade, nonmetastasizing, resectable osteosarcoma following complete surgical removal in children, adolescents, and young adults, aged two to 30 years.[1][6][7] Osteosarcoma is diagnosed in about 1,000 individuals in Europe and the USA per year, most under the age of 30.[8] The drug is used in combination with postoperative, multiagent chemotherapy to kill remaining cancer cells and improve a patient's chance of overall survival.[6]
In a phase-III clinical trial in about 800 newly diagnosed osteosarcoma patients, mifamurtide was combined with the chemotherapeutic agents doxorubicin and methotrexate, with or without cisplatin and ifosfamide. The mortality could be lowered by 30% versus chemotherapy plus placebo. Six years after the treatment, 78% of patients were still alive. This equals an absolute risk reduction of 8% .[1]
Adverse effects
In a clinical study, mifamurtide was given to 332 subjects (half of whom were under age of 16) and most side effects were found to be mild to moderate in nature. Most patients experience fewer adverse events with subsequent administration.[9][10]Common side effects include fever (about 90%), vomiting, fatigue and tachycardia (about 50%), infections, anaemia, anorexia, headache, diarrhoea and constipation(>10%).[1][11]
Pharmacokinetics
After application of the liposomal infusion, the drug is cleared from the plasma within minutes and is concentrated in lung, liver, spleen, nasopharynx, and thyroid. The terminal half-life is 18 hours. In patients receiving a second treatment after 11–12 weeks, no accumulation effects were observed.[12]
Pharmacodynamics
Mifamurtide is a fully synthetic derivative of muramyl dipeptide (MDP), the smallest naturally occurring immune stimulatory component of cell walls from Mycobacterium species. It has similar immunostimulatory effects as natural MDP with the advantage of a longer half-life in plasma.
NOD2 is a pattern recognition receptor which is found in several kinds of white blood cells, mainly monocytes and macrophages. It recognises muramyl dipeptide, a component of the cell wall of bacteria. Mifamurtide simulates a bacterial infection by binding to NOD2, activating white cells. This results in an increased production of TNF-α, interleukin 1,interleukin 6, interleukin 8, interleukin 12, and other cytokines, as well as ICAM-1. The activated white cells attack cancer cells, but not, at least in vitro, other cells.[13]
Interactions
- Theoretical considerations suggest calcineurin inhibitors like ciclosporin and tacrolimus might interact with mifamurtide because of their effect on macrophages.
- High-dose NSAIDs block the mechanism of mifamurtide in vitro.
Consequently, the combination of mifamurtide with these types of drugs is contraindicated. However, mifamurtide can be coadministered with low doses of NSAIDs. No evidence suggests mifamurtide interacts with the studied chemotherapeutics, or with the cytochrome P450 system.[14]
Chemistry

Mifamurtide is muramyl tripeptide phosphatidylethanolamine (MTP-PE), a synthetic analogue of muramyl dipeptide. The side chains of the molecule give it a longer elimination half-life than the natural substance. The substance is applied encapsulated into liposomes (L-MTP-PE). Being a phospholipid, it accumulates in the lipid bilayer of the liposomes in the infusion.[15]
Synthesis
One method of synthesis (shown first) is based on N,N'-dicyclohexylcarbodiimide (DCC) assisted esterification of N-acetylmuramyl-L-alanyl-D-isoglutaminyl-L-alanine with N-hydroxysuccinimide, followed by a condensation with 2-aminoethyl-2,3-dipalmitoylglycerylphosphoric acid in triethylamine (Et3N).[16] A different approach (shown second) uses N-acetylmuramyl-L-alanyl-D-isoglutamine, hydroxysuccinimide and alanyl-2-aminoethyl-2,3-dipalmitoylglycerylphosphoric acid;[17] that is, the alanine is introduced in the second step instead of the first.
 |  |
Synthesis
Mifamurtide is an anticancer agent for the treatment of osteosarcoma, the most common primary malignancy of bone tissue mainly affecting children and adolescents.10
The drug was invented by Ciba-Geigy (now Novartis) in the early 1980s and the agent was subsequently licensed to Jenner Biotherapies in the 1990s.
IDM Pharma bought the rights to the drug from Jenner in April 2003.78 In March 2009, mifamurtide was approved in the 27 European Union member states plus Iceland, Liechtenstein and Norway via a centralized marketing authorization.
After the approval, IDM Pharma was acquired by Takeda, which began launching mifamurtide, as Mepact , in February 2010.
Mifamurtide, a fully synthetic lipophilic derivative of muramyl dipeptide (MDP), is muramyl tripeptide phosphatidylethanolamine (MTP-PE), which is formulated as a liposomal infusion.79 Being a phospholipid, mifamurtide accumulates in the lipid bilayer of the liposomes upon infusion.
After application of the liposomal infusion, the drug is cleared from the plasma within minutes. However, it is concentrated in lung, liver, spleen, nasopharynx and thyroid, and the terminal half-life is 18 h, which is longer than the natural substance.
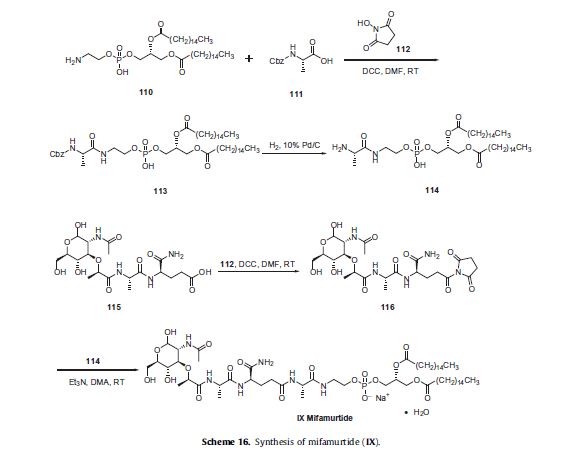
Two synthetic routes have been reported,80,81 and Scheme 16 describes the more processamenable route.
Commercially available 1,2-dipalmitoyl-sn-glycero- 3-phosphoethanolamine (110) was coupled with N-Boc-L-alanine (111) by means of N-hydroxysuccinimide (112), DCC in DMF to give amide 113, which was followed by hydrogenolysis of the CBZ group to give the corresponding L-alanyl-phosphoric acid 114.
Next, commercially available N-acetylmuramoyl-L-alanyl-Disoglutamine (115) was subjected to hydroxybenzotriazole (HOBT) and DIC in DMF to provide the corresponding succinimide ester 116 which was condensed with compound 114 to provide mifamurtide (IX).
No yields were provided for these transformations.
79. Prous, J. R.; Castaner, J. Drugs Future 1989, 14, 220.
80. Baschang, G.; Tarcsay, L.; Hartmann, A.; Stanek, J. EP 0027258 A1, 1980.
81. Brundish, D. E.; Wade, R. J. Labelled Compd. Radiopharm. 1985, 22, 29.
80. Baschang, G.; Tarcsay, L.; Hartmann, A.; Stanek, J. EP 0027258 A1, 1980.
81. Brundish, D. E.; Wade, R. J. Labelled Compd. Radiopharm. 1985, 22, 29.
PATENT
mifamurtide, the English called mifamurtide, formula C59Hltl9N6O19P, primarily for the treatment of non-metastatic
Resectable osteosarcoma (a rare but the main cause of death for children and young people osteoma), having the formula as follows:

mifamurtide by certain stimuli such as macrophages and other white blood cells to kill tumor cells. Currently, mifamurtide listed injections into spherical liposome vesicles are muramyl tripeptide (MTP). This lipid trigger macrophages to consume mifamurtide. Once consumed mifamurtide, MTP-stimulated macrophages, in particular we will look for tumors in the liver, spleen and lung macrophages and kill it.
mifamurtide injection approved for marketing based on the results of phase III clinical study. Taiwan's National Cancer Institute Cooperative Group (NCI) established by the Children's Oncology Group (COG) study, complete treatment of this product in patients with osteosarcoma largest research project in the book of about 800 cases. Evaluation of mifamurtide and 3-4 adjuvant chemotherapy (cis molybdenum, doxorubicin, methotrexate, cyclophosphamide with or the same as) the results of combination therapy. Studies have shown that mifamurtide used in combination with chemotherapy can reduce the mortality rate of about 30%, 78% of treated patients survived more than six years.
Shortcomings disclosed the full liquid phase synthesis technology route mifamurtide, but all-liquid phase synthesis: [0006] Currently, mifamurtide universal rely wholly liquid phase synthesis, relevant literature (220 Drugs Futl989, 14, (3)) that the synthesis requires intermediate purification steps cumbersome, time-consuming, and the total yield of the whole liquid phase synthesis is less than 30%, which has been the main factors affecting the productivity of mifamurtide
A method for logging meter synthetic peptide, characterized in that it comprises the following steps: Step 1, under the effect of coupling agent, an amino group, and Fmoc-D-Glu on the amino resin (OPG) -OH main chain carboxyl acylation, a compound of formula I; Step 2, Fmoc removal of the protecting group the compound of formula I, under the effect of coupling with Fmoc-L-Ala-OH acylation, a compound of formula 2; step 3, Fmoc removal of the protecting group the compound of formula 2, in the role of a coupling agent, with a compound of formula 3 for acylation, a compound of formula 4; step 4, PG protecting group removing compound of formula 4, the coupling the role of agent, and HL-Ala-OPG acylation, a compound of formula 5; Step 5, PG protecting group removal compound of formula 5, under the effect of coupling agent, and an amino acid performed on brain phospholipids reaction of a compound of formula 6, and then the resin was added Lysates deaminated compound of formula 7; Step 6, the compound of formula 7 to obtain the removal of benzyl mifamurtide;


Wherein Fmoc is the amino protecting group; wherein PG is a carboxy-protecting group for Allyl or Dmab; Resin as the amino resin.
Example: Synthesis of mifamurtide crude peptide
Example 11 to give the formula hydrogenolysis at atmospheric pressure to 16 hours Example 7 was added to 7.42 g compound 250ml single neck flask, dried 150ml of methanol was added to dissolve 0.4 g of 10% palladium on carbon.Completion of the reaction, palladium-carbon was filtered off, the filtrate was concentrated by rotary evaporation to 65ml, is mifamurtide crude peptide solution. Mifamurtide synthetic crude peptide: 15 [0173] Example
Example 12 to give the formula hydrogenolysis at atmospheric pressure to 16 hours Example 7 was added to 4.21 g compound 150ml single neck flask, dried 85ml of methanol was added to dissolve 0.2 g of 10% palladium on carbon.Completion of the reaction, palladium-carbon was filtered off, the filtrate was concentrated by rotary evaporation to 37ml, is mifamurtide crude peptide solution.
16 [0175] Example 2: Preparation of mifamurtide
The embodiment 14 of crude peptide solution obtained in Example 65ml, IOOOml round bottom flask was added, under magnetic stirring, 650ml of anhydrous diethyl ether was added dropwise. Upon completion, at room temperature for crystallization. After filtration and drying the filter cake, the filter cake was again dissolved in 65ml of methanol. This methanol solution was added IOOOml round bottom flask, under magnetic stirring, 650ml of anhydrous diethyl ether was added dropwise. Upon completion, at room temperature for crystallization. Filtered cake was dried in vacuo to give mifamurtide 5.62g, yield 86.5%, purity 99.4%, total yield 74.5%
Preparation of mifamurtide of: 17 Example
The embodiment of the crude peptide solution obtained in Example 15, 37ml, 500ml round bottom flask was added, under magnetic stirring, 370ml of anhydrous diethyl ether was added dropwise. Upon completion, at room temperature for crystallization. After filtration and drying the filter cake, the filter cake was again dissolved in 37ml of methanol. This solution was added to methanol 500ml round bottom flask, under magnetic stirring, 370ml of anhydrous diethyl ether was added dropwise. Upon completion, at room temperature for crystallization. Filtered, the filter cake was dried under vacuum to give · mifamurtide 3.16g, yield 85.8%, purity 99.5%, 72.2% overall yield.
References
- "Mifamurtide: CGP 19835, CGP 19835A, L-MTP-PE, liposomal MTP-PE, MLV 19835A, MTP-PE, muramyltripeptide phosphatidylethanolamine". Drugs in R&D 9 (2): 131–5. 2008. doi:10.2165/00126839-200809020-00007. PMID 18298131.
- "First Treatment to Improve Survival in 20 Years Now Available for Patients With Osteosarcoma (Bone Cancer)". Takeda. November 2009. Retrieved 23 March 2010.
- "IDM Pharma's MEPACT (Mifamurtide, L-MTP-PE) Receives Approval in Europe for Treatment of Patients with Non-Metastatic, Resectable Osteosarcoma". PR Newswire. 2009-03-09. Retrieved 2009-11-12.
- "IDM Pharma receives not approvable letter for Mifamurtide for treatment of osteosarcoma". The Medical News. 2007-08-28. Retrieved 2009-11-12.
- Mepact for Healthcare Professionals, retrieved 2009-11-12
- ^ Jump up to:a b EMA (2009-03-06). "Mepact: Product Information. Annex I: Summary of Product Characteristics" (PDF). p. 2. Retrieved 2009-11-12.
- EMA (2009-05-06). "Mepact: European Public Assessment Report. Summary for the public" (PDF). p. 1. Retrieved 2009-11-12.
- Meyers, P. A. (2009). "Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma". Expert Review of Anticancer Therapy 9 (8): 1035–1049.doi:10.1586/era.09.69. PMID 19671023.
- Meyers, P. A.; Schwartz, C. L.; Krailo, M. D.; Healey, J. H.; Bernstein, M. L.; Betcher, D.; Ferguson, W. S.; Gebhardt, M. C.; Goorin, A. M.; Harris, M.; Kleinerman, E.; Link, M. P.; Nadel, H.; Nieder, M.; Siegal, G. P.; Weiner, M. A.; Wells, R. J.; Womer, R. B.; Grier, H. E.; Children's Oncology, G. (2008). "Osteosarcoma: the Addition of Muramyl Tripeptide to Chemotherapy Improves Overall Survival--A Report from the Children's Oncology Group".Journal of Clinical Oncology 26 (4): 633–638. doi:10.1200/JCO.2008.14.0095.PMID 18235123.
- Meyers, P. A.; Schwartz, C. L.; Krailo, M.; Kleinerman, E. S.; Betcher, D.; Bernstein, M. L.; Conrad, E.; Ferguson, W.; Gebhardt, M.; Goorin, A. M.; Harris, M. B.; Healey, J.; Huvos, A.; Link, M.; Montebello, J.; Nadel, H.; Nieder, M.; Sato, J.; Siegal, G.; Weiner, M.; Wells, R.; Wold, L.; Womer, R.; Grier, H. (2005). "Osteosarcoma: A Randomized, Prospective Trial of the Addition of Ifosfamide and/or Muramyl Tripeptide to Cisplatin, Doxorubicin, and High-Dose Methotrexate". Journal of Clinical Oncology 23 (9): 2004–2011. doi:10.1200/JCO.2005.06.031. PMID 15774791.
- (EMA 2009, pp. 5–7)
- (EMA 2009, p. 8)
- (EMA 2009, pp. 7–8)
- (EMA 2009, p. 4)
- Fidler, I. J. (1982). "Efficacy of liposomes containing a lipophilic muramyl dipeptide derivative for activating the tumoricidal properties of alveolar macrophages in vivo". Journal of Immunotherapy 1 (1): 43–55.
- Prous, J. R.; Castaner, J. (1989). "ENV 2-3/MTP-PE". Drugs Fut. 14 (3): 220.
- Brundish, D. E.; Wade, R. (1985). "Synthesis of N-[2-3H]acetyl-D-muramyl-L-alanyl-D-iso-glutaminyl-L-alanyl-2-(1',2'-dipalmitoyl-sn-glycero-3'-phosphoryl)ethylamide of high specific radioactivity". J Label Compd Radiopharm 22 (1): 29–35. doi:10.1002/jlcr.2580220105.
| CN1055736A * | Jan 28, 1986 | Oct 30, 1991 | E·R·斯奎布父子公司 | Process for preparing 4,4-dialkyl-2-azetidinones |
| CN101709079A * | Dec 22, 2009 | May 19, 2010 | 江苏诺泰制药技术有限公司 | Synthesis method of romurtide |
| US4323560 * | Oct 6, 1980 | Apr 6, 1982 | Ciba-Geigy Corporation | Novel phosphorylmuramyl peptides and processes for the manufacture thereof |
| Reference | ||
|---|---|---|
| 1 | * | PROUS, J. ET AL: "ENV 2-3/MTP-PE", 《DRUGS FUT》, vol. 14, no. 3, 31 March 1989 (1989-03-31), pages 220 |
| 2 | * | 黄胜炎: "抗肿瘤药新品与研发进展", 《上海医药》, vol. 30, no. 9, 30 September 2009 (2009-09-30), pages 412 - 414 |
 | |
| Systematic (IUPAC) name | |
|---|---|
| 2-[(N-{(2R)-[(2-acetamido-2,3-dideoxy-D-glucopyranos-3-yl)oxy]-propanoyl}-L-alanyl-D-isoglutaminyl-L-alanyl)amino]ethyl (2R)-2,3-bis(hexadecanoyloxy)propyl hydrogen phosphate | |
| Clinical data | |
| License data | |
| Pregnancy category |
|
| Routes of administration | intravenous liposomal infusion over one hour |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | N/A |
| Biological half-life | minutes (in plasma) 18 hrs (terminal) |
| Identifiers | |
| CAS Number | 83461-56-7 838853-48-8 (mifamurtide sodium · xH2O) |
| ATC code | L03AX15 (WHO) |
| PubChem | CID 11672602 |
| ChemSpider | 9847332 |
| UNII | EQD2NNX741 |
| KEGG | D06619 |
| Chemical data | |
| Formula | C59H109N6O19P |
| Molar mass | 1237.499 g/mol |
//////////83461-56-7, 838853-48-8, CGP-19835, Mepact, MFCD09954133, Mifamurtide, mifamurtide sodium, MTP-cephalin, Mtp-PE, Muramyl tripeptide phosphatidylethanolamine, PEPTIDE, мифамуртид, ميفامورتيد, 米法莫肽
CCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1C(O)[C@@H](CO)O[C@@H](O)[C@@H]1NC(C)=O)C(N)=O)OC(=O)CCCCCCCCCCCCCCC