
IPI 504, Retaspamycin, Retaspimycin
CAS 857402-63-2
Cas 857402-23-4 ( Retaspimycin); 857402-63-2 ( Retaspimycin HCl).
MF C31H45N3O8 BASE
MW: 587.32067 BASE
Infinity Pharmaceuticals Inc, INNOVATOR
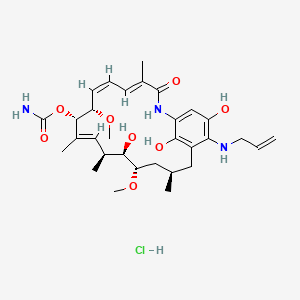
[(3R,5S,6R,7S,8E,10S,11S,12Z,14E)-6,20,22-trihydroxy-5,11-dimethoxy-3,7,9,15-tetramethyl-16-oxo-21-(prop-2-enylamino)-17-azabicyclo[16.3.1]docosa-1(22),8,12,14,18,20-hexaen-10-yl]
carbamate;hydrochloride17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride
| Retaspimycin hydrochloride; 8,21-didehydro-17-demethoxy-18,21-dideoxo-18,21-dihydroxy-17-(2-propenylamino)-geldanamycin monohydrochloride | |
| Application: | A novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90) |
| Molecular Weight: | 624.17 ..........HCl salt |
| Molecular Formula: | C31H46ClN3O8..........HCl salt |
Introduction
IPI-504 is a novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90).Orphan drug designation was assigned to the compound by the FDA for the treatment of gastrointestinal stromal cancer (GIST).

Retaspimycin Hydrochloride is the hydrochloride salt of a small-molecule inhibitor of heat shock protein 90 (HSP90) with antiproliferative and antineoplastic activities. Retaspimycinbinds to and inhibits the cytosolic chaperone functions of HSP90, which maintains the stability and functional shape of many oncogenic signaling proteins and may be overexpressed or overactive in tumor cells. Retaspimycin-mediated inhibition of HSP90 promotes the proteasomal degradation of oncogenic signaling proteins in susceptible tumor cell populations, which may result in the induction of apoptosis.
Phase I study of Retaspimycin: A phase 1 study of IPI-504 (retaspimycin hydrochloride) administered intravenously twice weekly for 2 weeks at 22.5, 45, 90, 150, 225, 300 or 400 mg/m(2) followed by 10 days off-treatment was conducted to determine the safety and maximum tolerated dose (MTD) of IPI-504 in patients with relapsed or relapsed/refractory multiple myeloma (MM). Anti-tumor activity and pharmacokinetics were also evaluated. Eighteen patients (mean age 60.5 years; median 9 prior therapies) were enrolled. No dose-limiting toxicities (DLTs) were reported for IPI-504 doses up to 400 mg/m(2).
The most common treatment-related adverse event was grade 1 infusion site pain (four patients). All other treatment-related events were assessed as grade 1 or 2 in severity. The area under the curve (AUC) increased with increasing dose, and the mean half-life was approximately 2-4 h for IPI-504 and its metabolites. Four patients had stable disease, demonstrating modest single-agent activity in relapsed or relapsed/refractory MM. (source: Leuk Lymphoma. 2011 Dec;52(12):2308-15.)

In a different approach, Infinity Pharmaceuticals
has developed IPI504 (retaspimycin or 17-AAG hydroquinone, Figure 4)
(Adams et al., 2005; Sydor et al., 2006), a new GA analogue, in which
the quinone moiety was replaced by a dihydroquinone one. Indeed, the
preclinical data suggested that the hepatotoxicity of 17-AAG was
attributable to the ansamycin benzoquinone moiety, prone to nucleophilic
attack.
Furthermore, it was recently reported
that the hydroquinone form binds Hsp90 with more efficiency than the
corresponding quinone form (Maroney et al., 2006). In biological
conditions, the hydroquinone form can interconvert with GA, depending on
redox equilibrium existing in cell. It has been recently proposed, that
NQ01 (NAD(P)H: quinone oxidoreductase) can produce the active hydroquinone from the quinone form of IPI504 (Chiosis, 2006).
However, Infinity Pharmaceuticals
showed that if the overexpression of NQ01 increased the level of
hydroquinone and cell sensitivity to IPI504, it has no significant
effect on its growth inhibitory activity. These results suggest that
NQ01 is not a determinant of IPI504 activity in vivo (Douglas et al., 2009).

PATENT
http://www.google.com/patents/EP2321645A1?cl=enGeldanamycin (IUPAC name ([18S-(4E,5Z,8R*.9R*.10E,12R*.13S*,14R*,l6S*)]- 9- [(aminocarbonyl)oxy]- 13- hydroxy- 8,14,19- trimetoxy- 4,10,12,16- tetramethyl- 2- azabicyclo[16.3.1.]docosa- 4,6.10,18,21- pentan- 3.20,22trion) is a benzoquinone ansamycin antibiotic which may be produced by the bacterium Streptorayces hygroscopicus. Geldanamycin binds specifically to HSP90 (Heat Shock Protein 90) and alters its function.
While Hsp90 generally stabilizes folding of proteins and, in particular in tumor cells, folding of overexpressed/mutant proteins such as v-Src. Bcr-Abl and p53. the Hsp90 inhibitor Geldanamycin induces degradation of such proteins.
The respectiv e formula of geldanamycin is given herein below:

Accordingly geldanamycin analogues/derivatives which are modified at the 17 position, such as 17-AAG (17-N-Allylamino-17-demethoxygeldanamycin), are preferred in context of the present invention. Also preferred herein are geldanamycin derivatives to be used in accordance with the present invention which are water-soluble or which can be dissoh ed in water completely (at least 90 %. more preferably 95 %. 96 %. 97 %, 98 % and most preferably 99 %). 17-AAG ([QS.5S,6RJS$EΛ0R,l \SΛ2E,14E)-2\- (allylamino)-6-hydroxy-5.11-diraethoxy-
3.7.9,15-tetramethyl-16.20.22-trioxo-17-azabicyclo[16.3.1]docosa-8,12.14,18,21-pentaen-10- yl] carbamate) is. as mentioned above a preferred derivative of geldanamycin. 17- AAG is commercially available under the trade name "Tanespimycin" (also known as KOS-953) for example from Kosan Biosciences Incorporated (Acquired by Bristol-Myers Squibb Company). Tanespimycin is presently studied in phase II clinical trials for multiple myeloma and breast cancer and is usually administered intravenously.
The respective formula of 17- AAG is given herein below:


PATENT
WO 2005063714http://www.google.co.ug/patents/WO2005063714A1?cl=en
Example 24
Preparation of Air-stable Hydroquinone Derivatives of the Geldanamycin Family of Molecules
,

(200 mL) was stirred vigorously with a freshly prepared solution of 10% aqueous sodium hydrosulfite (200 mL) for 2 h at ambient temperature. The color changed from dark purple to bright yellow, indicating a complete reaction. The layers were separated and the organic phase was dried with magnesium sulfate (15 g). The drying agent was rinsed with ethyl acetate (50 mL). The combined filtrate was acidified with 1.5 M hydrogen chloride in ethyl acetate (12 mL) to pH 2 over 20 min. The resulting slurry was stirred for 1.5 h at ambient temperature. The solids were isolated by filtration, rinsed with ethyl acetate (50 mL) and dried at 40 °C, 1 mm Hg, for 16 h to afford 9.9 g (91%) of off-white solid. Crude hydroquinone hydrochloride (2.5 g) was added to a stirred solution of 5% 0.01 N aq. hydrochloric acid in methanol (5 mL). The resulting solution was clarified by filtration then diluted with acetone (70 mL). Solids appeared after 2-3 min. The resulting slurry was stirred for 3 h at ambient temperature, then for 1 h at 0-5 °C. The solids were isolated by filtration, rinsed with acetone (15 mL) and dried
PAPER
J. Med. Chem., 2006, 49 (15), pp 4606–4615
DOI: 10.1021/jm0603116

17-Allylamino-17-demethoxygeldanamycin (17-AAG)1
is a semisynthetic inhibitor of the 90 kDa heat shock protein (Hsp90)
currently in clinical trials for the treatment of cancer. However,
17-AAG faces challenging formulation issues due to its poor solubility.
Here we report the synthesis and evaluation of a highly soluble
hydroquinone hydrochloride derivative of 17-AAG, 1a (IPI-504),
and several of the physiological metabolites. These compounds show
comparable binding affinity to human Hsp90 and its endoplasmic reticulum
(ER) homologue, the 94 kDa glucose regulated protein (Grp94).
Furthermore, the compounds inhibit the growth of the human cancer cell
lines SKBR3 and SKOV3, which overexpress Hsp90 client protein Her2, and
cause down-regulation of Her2 as well as induction of Hsp70 consistent
with Hsp90 inhibition. There is a clear correlation between the measured
binding affinity of the compounds and their cellular activities. Upon
the basis of its potent activity against Hsp90 and a significant
improvement in solubility, 1a is currently under evaluation in Phase I clinical trials for cancer.
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride Ia
17-AAG hydroquinone hydrochloride (1a) as an off-white solid (11 g, 18 mmol, 80% yield). HPLC purity: 99.6%;IR (neat): 3175, 2972, 1728, 1651, 1581, 1546, 1456, 1392, 1316, 1224, 1099, 1036 cm-1;
1H NMR (CDCl3:d6-DMSO, 6:1, 400 MHz):
δ 10.20 (1H, br), 9.62 (2H, br), 8.53 (1H, s), 8.47 (1H, s), 7.74 (1H, s), 6.72 (1H, d, J= 11.6 Hz), 6.28 (1H, dd, J = 11.6, 11.2 Hz), 5.73 (1H, dddd, J = 17.2, 10.0, 3.2, 2.4 Hz), 5.53 (1H, d, J = 10.4 Hz), 5.49 (1H, dd, J = 10.8, 10.0 Hz), 5.32 (2H, s), 5.04 (1H, d, J = 4.8 Hz), 5.02 (1H, d, J = 16.0 Hz), 4.81 (1H, s), 4.07 (1H, d, J = 9.6 Hz), 3.67 (2H, d, J = 6.4 Hz), 3.31 (1H, d,J = 8.8 Hz), 3.07 (3H, s), 3.07−3.04 (1H, m), 2.99 (3H, s), 2.64 (1H, m), 2.52−2.49 (1H, m), 1.76 (3H, s), 1.61−1.39 (3H, m), 0.78 (3H, d, J = 6.4 Hz), 0.64 (3H, d, J = 7.2 Hz);
13C NMR (CDCl3:d6-DMSO, 6:1, 100 MHz): δ 167.3, 155.8, 143.3, 136.3, 135.0, 134.2, 132.9, 132.1, 128.8, 127.6, 125.9, 125.3, 123.7, 123.0, 115.1, 104.5, 80.9, 80.7, 80.1, 72.5, 56.2, 56.2, 52.4, 34.6, 33.2, 31.1, 27.2, 21.6, 12.1, 12.1, 11.7;
HRMS calculated for C31H45N3O8 (M+ + H): 588.3285, Found 588.3273.
POSTER
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
MEDI 210 |
| IPI-504
is the hydroquinone hydrochloride salt of
17-allylamino-17-demethoxy-geldanamycin (17-AAG), an Hsp90 inhibitor
that is currently in clinical trials for the treatment of cancer. IPI-504 demonstrates high aqueous solubility (>200 mg/mL). Interestingly, in vitro and in vivo IPI-504 interconverts with 17-AAG and exists in a pH and enzyme-mediated redox equilibrium. This occurs due to oxidation of the hydroquinone (IPI-504) to the quinone (17-AAG) at physiological pH and the reduction of 17-AAG by quinone reductases such as NQO1 to IPI-504. Here we report the design and synthesis of the stabilized hydroquinone IPI-504 and its inhibitory effect against Hsp90 and Grp94. Although IPI-504 was originally designed to be a soluble prodrug of 17-AAG, the hydroquinone is more potent than the quinone in the biochemical Hsp90 binding assay. Various hydroquinone analogs have been prepared to investigate the structure activity relationship of hydroquinone binding to Hsp90. Hydroquinone and quinone forms of 17-AAG metabolites show comparable binding affinities for Hsp90 and in cancer cell lines, hydroquinone analogs elicit specific responses consistent with Hsp90 inhibition. The desirable pharmacological properties as well as in vitro and in vivo activity of our lead compound, IPI-504, has led to the initiation of Phase I clinical trials in multiple myeloma. |
| http://oasys2.confex.com/acs/231nm/techprogram/P945016.HTM |
Geldanamycin and Beyond: Progress Toward the Development of HSP90 Inhibitors
8:30 AM-12:05 PM, Wednesday, 29 March 2006 Georgia World Congress Center -- Georgia Ballroom 1, OralDivision of Medicinal ChemistryThe 231st ACS National Meeting, Atlanta, GA, March 26-30, 2006 |
References
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90231st Am Chem Soc (ACS) Natl Meet (March 26-30, Atlanta) 2006, Abst MEDI 210
Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90
J Med Chem 2006, 49(15): 4606
http://www.biotechduediligence.com/retaspamycin-hcl-ipi-504.html
///////////////////Hsp90, IPI-504, infinity pharma, Retaspamycin, Retaspimycin



![ChemSpider 2D Image | N-[(2S,3R,3'R,3aR,4a'R,6S,6a'R,6b'S,7aR,12a'S,12b'S)-3,6,11',12b'-Tetramethyl-2',3',3a,4,4',4a',5,5',6,6',6a',6b',7,7',7a,8',10',12',12a',12b'-icosahydro-1'H,3H-spiro[furo[3,2-b]pyridine-2,9'-naphtho[ 2,1-a]azulen]-3'-yl]methanesulfonamide | C29H48N2O3S](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_ucUK9EAecyha4DUWoCGkJwonovyHHL0izygPuOO6F5Tc15F_U89OgpN67ms71CsTh4H08EMLqFCZHIjFme6RctKZ4ZMyQnvBSVk7IfYEV5U3OyPGrU4UIifZkinovkcI_yVRxkNPA0=s0-d)

















 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPD












