

Nacubactam
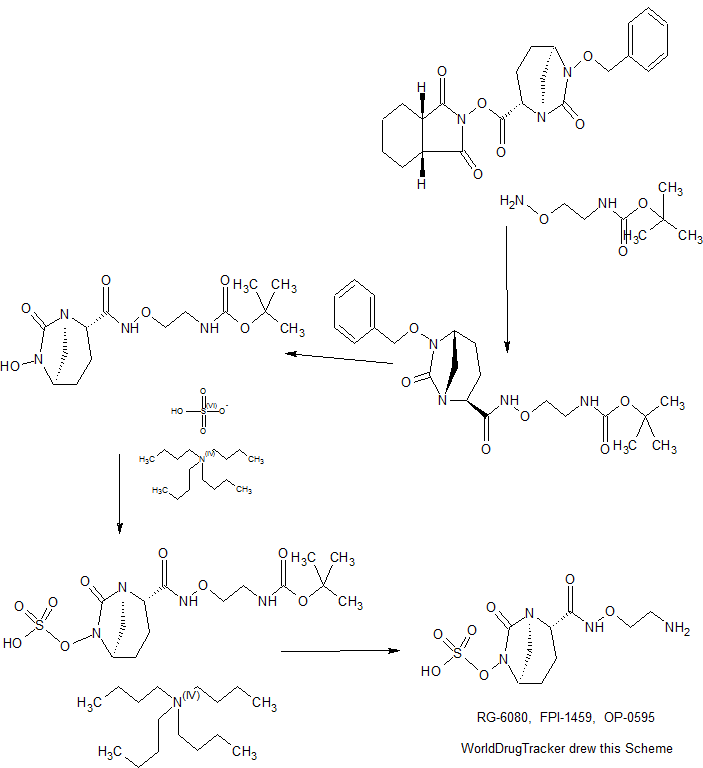
RG-6080, FPI-1459, OP-0595, WK ?, WK-?, WK?
CAS 1452458-86-4, MF C9 H16 N4 O7 S, MW 324.31
Sulfuric acid, mono[(1R,2S,5R)-2-[[(2-aminoethoxy)amino]carbonyl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester,
(2S,5R)-N-(2-amino ethoxy)-6-(sulfooxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
Beta lactamase inhibitor
Roche, under license from Meiji Seika Pharma and Fedora Pharmaceuticals is developing nacubactam hydrate
A diazabicyclooctane beta-lactamase inhibitor, for treating bacterial infection. In July 2016, nacubactam was reported to be in phase 1 clinical development
PATENTS , IN2015MU287, WO2016116878, WO 2016120752, INDICATE INTEREST FROM WOCKHARDT


Sulfuric acid, mono[(1R,2S,5R)-2-[[(2-aminoethoxy)amino]carbonyl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
A β-lactamase inhibitor potentially for the treatment of bacterial infections.
RG-6080; FPI-1459; OP-0595
CAS No. 1452458-86-4
| Molecular Formula | C9 H16 N4 O7 S |
| Formula Weight | 324.31 |
- Originator Fedora Pharmaceuticals
- Developer Meiji Seika Pharma
- Class Antibacterials; Azabicyclo compounds
- Mechanism of Action Beta lactamase inhibitors
- Phase I Bacterial infections
Most Recent Events
- 13 Jan 2015 OP 0595 licensed to Roche worldwide, except Japan ,
- 30 Nov 2014 Meiji Seika Pharma completes a phase I trial in Healthy volunteers in Australia (NCT02134834)
- 01 May 2014 Phase-I clinical trials in Bacterial infections (in volunteers) in Australia (IV)

In September 2014, preclinical data were presented at the 54th ICAAC Meeting in Washington, DC. Nacubactam hydratedemonstrated Ki values of 0.24, 3 and 0.79 microM against AmpC P99 derived from Enterobacter cloacae, KPC-3, and CTX-M-15 enzymes, respectively; the Ki values were lower than that of cefepime
Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate.
The persistent exposure of bacterial strains to a multitude of beta- lactam antibacterial agents has led to overproduction and mutation of beta-lactamases. These new extended spectrum beta-lactamases (ESBL) are capable of hydrolyzing penicillins, cephalosporins, monobactams and even carbapenems. Such a wide spread resistance to many of the existing beta-lactam antibacterial agents, either used alone or in combination with other agents, is posing challenges in treating serious bacterial infections.
Due to various reasons, the oral therapeutic options for treating bacterial infections (including those caused by ESBL strains) are limited. For example, a combination of amoxicillin and clavulanic acid is effective against Class A ESBLs producing bacteria. However, the usefulness of this combination is compromised against bacteria producing multiple or mixed beta-lactamase enzymes (such as, for example, bacteria producing Class A and Class C ESBLs concurrently), and Klebsiella pneumoniae carbapenemases (KPCs). Therefore, oral antibacterial agents or combinations with activity against a range of bacterial strains (including those producing multiple ESBLs and KPCs) are urgently desired.
Cephalosporin antibacterial agents are known for treatment for various bacterial infections. Surprisingly, it has been found that pharmaceutical compositions comprising a cephalosporin antibacterial agent and certain nitrogen containing bicyclic compound (disclosed in PCT/IB2013/053092, PCT/JP2013/064971 and PCT/IB 2012/002675) exhibit unexpectedly synergistic antibacterial activity, even against highly resistant bacterial strains.
The persistent exposure of bacterial strains to a multitude of beta- lactam antibacterial agents has led to overproduction and mutation of beta-lactamases. These new extended spectrum beta-lactamases (ESBL) are capable of hydrolyzing penicillins, cephalosporins, monobactams and even carbapenems. Such a wide spread resistance to many of the existing beta-lactam antibacterial agents, either used alone or in combination with other agents, is posing challenges in treating serious bacterial infections.
Due to various reasons, the oral therapeutic options for treating bacterial infections (including those caused by ESBL strains) are limited. For example, a combination of amoxicillin and clavulanic acid is effective against Class A ESBLs producing bacteria. However, the usefulness of this combination is compromised against bacteria producing multiple or mixed beta-lactamase enzymes (such as, for example, bacteria producing Class A and Class C ESBLs concurrently), and Klebsiella pneumoniae carbapenemases (KPCs). Therefore, oral antibacterial agents or combinations with activity against a range of bacterial strains (including those producing multiple ESBLs and KPCs) are urgently desired.
Cephalosporin antibacterial agents are known for treatment for various bacterial infections. Surprisingly, it has been found that pharmaceutical compositions comprising a cephalosporin antibacterial agent and certain nitrogen containing bicyclic compound (disclosed in PCT/IB2013/053092, PCT/JP2013/064971 and PCT/IB 2012/002675) exhibit unexpectedly synergistic antibacterial activity, even against highly resistant bacterial strains.
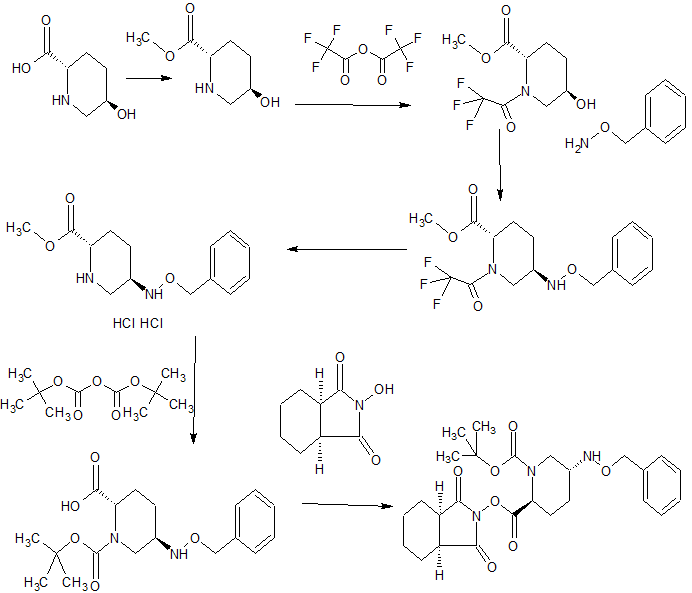
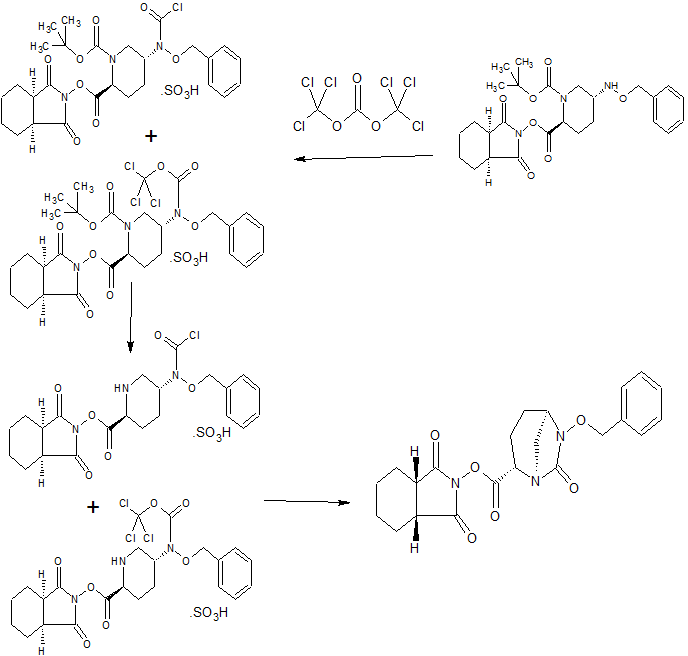
SYNTHESIS
WO 2015046207,
CONTD.......................
CONTD......................................
Patent
The novel heterocyclic compound in Japanese Patent 4515704 (Patent Document 1), preparation and shown for their pharmaceutical use, sodium trans-7-oxo-6- (sulfooxy) as a representative compound 1,6-diazabicyclo [3 .2.1] discloses an octane-2-carboxamide (NXL104). Preparation in regard to certain piperidine derivatives which are intermediates Patent 2010-138206 (Patent Document 2) and JP-T 2010-539147 (Patent Document 3) are shown at further WO2011 / 042560 (Patent Document 4) NXL104 to disclose a method for producing the crystals.
In Patent 5038509 (Patent Document 5) (2S, 5R) -7- oxo -N- (piperidin-4-yl) -6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane - 2- carboxamide (MK7655) is shown, discloses the preparation of certain piperidine derivatives with MK7655 at Patent 2011-207900 (Patent Document 6) and WO2010 / 126820 (Patent Document 7).
The present inventors also disclose the novel diazabicyclooctane derivative represented by the following formula (VII) in Japanese Patent Application 2012-122603 (Patent Document 8).
Patent Document 1: Japanese Patent No. 4515704 Pat
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
[Chemical formula 1] (In the formula, R 3 are the same as those described below)

Reference Example
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
step 1 tert-butyl {2 - [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate (IV-1)(2S, 5R)-6-(benzyloxy) -7-oxo-1,6-diazabicyclo [3.2.1] octane-2-carboxylic acid (4 .30g, dehydrated ethyl acetate (47mL) solution of 15.56mmol) was cooled to -30 ℃, isobutyl chloroformate (2.17g, washing included dehydration ethyl acetate 1mL), triethylamine (1.61g, washing included dehydration ethyl acetate 1 mL), successively added dropwise, and the mixture was stirred 1 hour at -30 ° C.. To the reaction solution tert- butyl 2-dehydration of ethyl acetate (amino-oxy) ethyl carbamate (3.21g) (4mL) was added (washing included dehydration ethyl acetate 1mL), raising the temperature over a period of 1.5 hours to 0 ℃, It was further stirred overnight. The mixture of 8% aqueous citric acid (56 mL), saturated aqueous sodium bicarbonate solution (40 mL), sequentially washed with saturated brine (40 mL), dried over anhydrous magnesium sulfate, filtered, concentrated to 5 mL, up to 6mL further with ethanol (10 mL) It was replaced concentrated. Ethanol to the resulting solution (3mL), hexane the (8mL) in addition to ice-cooling, and the mixture was stirred inoculated for 15 minutes. The mixture was stirred overnight dropwise over 2 hours hexane (75 mL) to. Collected by filtration the precipitated crystals, washing with hexane to give the title compound 5.49g and dried in vacuo (net 4.98 g, 74% yield). HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 50/50, 1.0 mL / min, UV 210 nm, Retweeted 4.4 min; 1 H NMR (400 MHz, CDCl 3 ) [delta] 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m, 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br.d., J = 11.6 Hz , 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br. . s, 1H), 7.34-7.48 (yd, 5H), 9.37 (Br.S., 1H); MS yd / z 435 [M + H] + .

Step 2
tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate
(V-1) tert-butyl {2 - [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
(V-1) tert-butyl {2 - [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
Step 3
Tetrabutylammonium tert- butyl {2 - [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide - pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
Tetrabutylammonium tert- butyl {2 - [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide - pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
Step 4 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
tetra butylammonium tert- butyl {2 - [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
tetra butylammonium tert- butyl {2 - [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
PATENT
Example
64 tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]

64 tert-butyl {2 - [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]
tert- butyl {2 - [({[(2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate (example 63q, net 156.42g, 360mmol) in methanol solution (2.4L) of 10% palladium carbon catalyst (50% water, 15.64g) was added, under an atmosphere of hydrogen, stirred for 1.5 hours did. The catalyst was filtered through celite, filtrate was concentrated under reduced pressure until 450mL, concentrated to 450mL by adding acetonitrile (1.5 L), the mixture was stirred ice-cooled for 30 minutes, collected by filtration the precipitated crystals, washing with acetonitrile, and vacuum dried to obtain 118.26g of the title compound (net 117.90g, 95% yield). Equipment data of the crystals were the same as those of the step 2 of Reference Example 3.
Example
65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)

65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)
tert- butyl {2 - [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (example 64,537.61g, 1.561mol) in acetonitrile (7.8L) solution of 2,6-lutidine (512.08g), sulfur trioxide - pyridine complex (810.3g) was added, at room temperature in the mixture was stirred overnight. Remove insolubles and the mixture was filtered, the filtrate concentrated to 2.5 L, diluted with ethyl acetate (15.1L). The mixture was extracted with 20% phosphoric acid 2 hydrogencarbonate aqueous solution (7.8L), the resulting aqueous layer into ethyl acetate (15.1L), added tetrabutylammonium hydrogen sulfate (567.87g), was stirred for 20 min. The organic layer was separated layers, dried over anhydrous magnesium sulfate (425 g), after filtration, concentration under reduced pressure, substituted concentrated tetrabutylammonium tert- butyl with dichloromethane (3.1L) {2 - [({[(2S, 5R ) -7-oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 758g (net 586.27g, Osamu rate 84%).
The tetra-butyl ammonium salt 719g (net 437.1g, 0.656mol) in dichloromethane (874mL) solution was cooled to -20 ℃, dropping trifluoroacetic acid (874mL) at 15 minutes, 1 the temperature was raised to 0 ℃ It was stirred time. The reaction was cooled to -20 ° C. was added dropwise diisopropyl ether (3.25L), and the mixture was stirred for 1 hour the temperature was raised to 0 ° C.. The precipitate is filtered, washed with diisopropyl ether to give the title compound 335.36g of crude and vacuum dried (net 222.35g, 99% yield).
The title compound of crude were obtained (212.99g, net 133.33g) and ice-cold 0.2M phosphate buffer solution of pH5.3 mix a little at a time, alternating between the (pH6.5,4.8L). The solution was concentrated under reduced pressure to 3.6L, it was adjusted to pH5.5 at again 0.2M phosphate buffer (pH6.5,910mL). The solution resin purification (Mitsubishi Kasei, SP207, water ~ 10% IPA solution) is subjected to, and concentrated to collect active fractions, after lyophilization, to give the title compound 128.3 g (96% yield). Equipment data of the crystals were the same as those of step 3 of Reference Example 3.
PATENT
US 20140288051
WO 2014091268
WO 2013180197
US 20130225554
PATENT
IN2015MU287
PATENT
Example 59
(2S, 5R) -N- (2- aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (II-059)
(2S, 5R) -N- (2- aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (II-059)

Step 1
tert- butyl {2 - [({[(2S, 5R) -6- Benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate
tert- butyl {2 - [({[(2S, 5R) -6- Benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate

Acid of Example 9 or 16 (6b, 1.34g, 4.87mmol) in methylene chloride (35mL) solution of triethylamine (2.71mL), N- ethyl -N '- (3- dimethylaminopropyl) carbodiimide hydrochloride (1.41g), 1- hydroxybenzotriazole monohydrate (1.15g), were added tert- butyl of Reference Example 9, wherein 2- (amino-oxy) ethyl carbamate (1.12g), room temperature It was stirred overnight Te.Water was added to the reaction solution to a residue obtained by concentration under reduced pressure, and extracted with ethyl acetate. The resulting organic layer with 0.1M hydrochloric acid, saturated aqueous sodium bicarbonate solution, washed with saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered and concentrated.The resulting residue was purified by silica gel column and purified by chromatography (hexane / ethyl acetate = 8 / 2-0 / 10) to give the title compound 1.77g (84% yield).
[Α] D 20 -0.08 ° (c 0.29, CHCl 3); 1 H NMR (400 MHz, CDCl 3), δ: 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m , 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br d, J = 11.6 Hz, 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br s, 1H), 7.34-7.48 (m, 5H), 9.37 (br s, 1H); MS m / z 435 [M + H] +; enantiomeric excess of 99.9% or higher ee (CHIRALPAK AD-H, 4.6x150mm, hexane / ethanol = 2/1, UV210nm, flow rate 1mL / min, retention time 4.95min (2R, 5S), 6.70min (2S, 5R).
[Α] D 20 -0.08 ° (c 0.29, CHCl 3); 1 H NMR (400 MHz, CDCl 3), δ: 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m , 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br d, J = 11.6 Hz, 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br s, 1H), 7.34-7.48 (m, 5H), 9.37 (br s, 1H); MS m / z 435 [M + H] +; enantiomeric excess of 99.9% or higher ee (CHIRALPAK AD-H, 4.6x150mm, hexane / ethanol = 2/1, UV210nm, flow rate 1mL / min, retention time 4.95min (2R, 5S), 6.70min (2S, 5R).
Step 2
tert- butyl {2 - [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate
tert- butyl {2 - [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate

Compound of the above Step 1 (3.91g, 9.01mmol) in methanol (80mL), 10% palladium on carbon catalyst (50% water, 803mg) was added, under hydrogen atmosphere and stirred for 45 minutes. The reaction mixture was filtered through Celite, then concentrated under reduced pressure, to give 3.11g of the title compound (quantitative).
1 H NMR (400 MHz, CD 3 OD), δ: 1.44 (s, 9H), 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 ( br dd, J = 15.0, 7.0 Hz, 1H), 3.03 (d, J = 12.0 Hz, 1H), 3.12 (br d, J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS m / z 345 [M + H] +.
1 H NMR (400 MHz, CD 3 OD), δ: 1.44 (s, 9H), 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 ( br dd, J = 15.0, 7.0 Hz, 1H), 3.03 (d, J = 12.0 Hz, 1H), 3.12 (br d, J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS m / z 345 [M + H] +.
Step 3
(2S, 5R) -N- (2- aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide The above step 2 compound (3. 09g, in methylene chloride (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide - was added pyridine complex (3.58g), and stirred at room temperature overnight. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, and washed the aqueous layer with chloroform, and tetrabutylammonium hydrogen sulfate (3.47g) and chloroform (30mL) was added to the aqueous layer and stirred for 10 minutes. After extracting the aqueous layer with chloroform, drying the resulting organic layer over anhydrous sodium sulfate, filtered, concentrated under reduced pressure tetrabutylammonium tert- butyl {2 - [({[(2S, 5R) -7- oxo - 6- (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 5.46g (91% yield).
1 H NMR (400 MHz, CDCl 3), δ: 1.01 (t, J = 7.4 Hz, 12H), 1.37-1.54 (m, 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30-2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85- 3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s, 1H), 9.44 (br s, 1H); MS m / z 425 [M-Bu 4 N + 2H] +.
(2S, 5R) -N- (2- aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide The above step 2 compound (3. 09g, in methylene chloride (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide - was added pyridine complex (3.58g), and stirred at room temperature overnight. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, and washed the aqueous layer with chloroform, and tetrabutylammonium hydrogen sulfate (3.47g) and chloroform (30mL) was added to the aqueous layer and stirred for 10 minutes. After extracting the aqueous layer with chloroform, drying the resulting organic layer over anhydrous sodium sulfate, filtered, concentrated under reduced pressure tetrabutylammonium tert- butyl {2 - [({[(2S, 5R) -7- oxo - 6- (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 5.46g (91% yield).
1 H NMR (400 MHz, CDCl 3), δ: 1.01 (t, J = 7.4 Hz, 12H), 1.37-1.54 (m, 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30-2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85- 3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s, 1H), 9.44 (br s, 1H); MS m / z 425 [M-Bu 4 N + 2H] +.
The tetrabutyl ammonium salt (5.20g, 7.82mmol) in methylene chloride (25mL) solution of under ice-cooling trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed resulting residue with diethyl ether, at aqueous sodium bicarbonate was adjusted to pH7, it performs an octadecyl silica gel column chromatography (water), after freeze-drying, 1.44g of the title compound The obtained (57% yield).
[Α] D 24 -63.5 ° (c 0.83, H 2 O); 1 H NMR (400 MHz, D 2 O), δ: 1.66-1.76 (m, 1H), 1.76-1.88 (m, 1H), 1.91 -2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz, 2H), 3.18 (br d, J = 12.0 Hz , 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4, 3.2 Hz, 1H); MS m / z 325 [ M + H] +.
[Α] D 24 -63.5 ° (c 0.83, H 2 O); 1 H NMR (400 MHz, D 2 O), δ: 1.66-1.76 (m, 1H), 1.76-1.88 (m, 1H), 1.91 -2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz, 2H), 3.18 (br d, J = 12.0 Hz , 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4, 3.2 Hz, 1H); MS m / z 325 [ M + H] +.
PATENT

ANTIBACTERIAL COMPOSITIONS OF A BETA-LACTAMASE INHIBITOR WITH A CEPHALOSPORINAbstract:
Pharmaceutical compositions comprising: (a) at least one cephalosporin antibacterial agent and (b) a compound of Formula (I) or a stereoisomer or a pharmaceutically acceptable derivative thereof are disclosed. Formula (I)
PATENT
WO 2016120752, WOCKHARDT, NEW PATENT, Nacubactam

Formula (I), chemically known as (25, 5i?)-N-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide has antibacterial properties and is disclosed in PCT International Patent Application No. PCT/IB2013/053092, PCT/JP2013/064971 and PCT/IB2012/002675. The present invention discloses a process for preparation of a compound of Formula (I).
Formula (I)

(VII) (VIII) (IX)
Scheme 2
Example 1
Synthesis of fert-butyl-r2-(aminooxy) ethyllcarbamate (III)
Preparation of fert-butyl-2-hydroxy ethylcarbamate (VIII):
Formula (VIII)

To a stirred solution of ethanolamine (50.0 g, 0.8186 mol) in dichloromethane (1000 ml), was added triethylamine (124 g, 1.228 mol) at 0°C. After 10 minutes, di-teri-butyl dicarbonate (VII, 214.15 g, 0.9823 mol) was added drop wise at 0°C under continuous stirring. Then reaction mass was allowed to warm to 25°C and stirred further for 3 hours. After completion of reaction, the resulting reaction mixture was poured into water (250 ml) and the organic layer was separated and dried over anhydrous sodium sulfate. The dried organic layer was concentrated under reduced pressure to obtain 130 g of the titled product as colorless oil in 98% yield.
Analysis:
Mass: 162 (M+l); for Molecular Weight of 161.2 and Molecular Formula of C7H15NO3.
1H NMR (400MHz, CDC13): δ 4.92(br s,lH), 3.72-3.68(q,2H), 3.30-3.26(q,2H), 2.33(br s,lH), 1.44(s,9H).
Preparation of A7-Boc-2-(2-aminoethoxy)isoindoline-l,3-dione (IX):

To a stirred solution of teri;butyl-2-hydroxy-ethylcarbamate (VIII, 50 g, 0.3106 mol) in tetrahydrofuran (500 ml), was added triphenylphosphine (89.5 g, 0.3416 mol) at 25°C. After stirring for 10 minutes, a solution of N-hydroxyphthalimide (50.66 g, 0.3106 mol) in dichloromethane (250 ml) was added to the reaction mass at 25 °C over a period of 10 minutes. After stirring for further 10 minutes, diisopropyl azodicarboxylate (69.1 g, 0.3416 mol) was added to the reaction mass in small portions (exothermic reaction was observed up to 34°C). The resulting reaction mass was stirred further at 25°C. After 16 hours, the reaction mass was concentrated under reduced pressure to obtain colorless oily material. The oily residue was diluted with diisopropyl ether (200 ml) and stirred for 30 minutes. The separated solid was filtered under suction. The filtrate was evaporated under reduced pressure and the residue subjected to di-isopropyl ether treatment (200 ml). This procedure was repeated once again. The filtrate was concentrated to obtain a solid product. The obtained solid was washed with diisopropyl ether (50 ml) and dried under reduced pressure. This solid contains small amount of triphenylphosphine oxide, along with the product. This was used as such for the next reaction without further purification.
Analysis:
Mass: 307.2 (M+l); for Molecular Weight of 306.3 and Molecular Formula of Ci5Hi8N205; 1H NMR of purified material (400MHz, CDC13): 7.85-7.25 (m,4H), 5.62(br s,lH), 4.26-4.23(t,2H), 3.46-3.42(q,2H), 1.46(s,9H).
Step 3: Preparation of fert-butyl-[ -(aminooxy) ethyl]carbamate (III):
Formula (III)

To a stirred solution of N-Boc-2-(2-aminoethoxy)isoindoline-l ,3-dione (IX, 97 g, 0.3167 mol) in dichloromethane (970 ml) was added hydrazine hydrate (31.7 g, 0.6334 mol) , at 0°C, drop wise, over a period of 45 minutes and the stirring continued further. After 2 hours, the reaction mass was filtered under suction. Filtrate was washed with water (485 ml), and the organic layer was diluted with an aq. solution of 10% potassium hydrogen sulfate (485 ml) and stirred for 15 minutes. The aqueous layer was separated, neutralized with solid sodium hydrogen carbonate and extracted with dichloromethane (2 x 485 ml). The organic layer was separated, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain colorless oil, this was used as such for further reaction immediately (28g, overall yield of step II and step III was 60%)
Analysis:
Mass: 177.2 (M+l) for Molecular Weight of 176.2 and Molecular Formula of C7H16N2O3.
Example 2
Synthesis of (25,5R)-jV-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicvclor3.2.11octane-2- carboxamide (I)
Step 1: Preparation of (25,5R)-iV-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (IV):

To a clear solution of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II, 42.67 g, 0.143 mol; prepared according to the procedure disclosed in Indian Patent Application No. 699/MUM/2013) in water (426 ml) was added EDC.HC1 (67.1 g, 0.349 mol) at 15°C
under stirring. After 10 minutes, a solution of teri-butyl-[2-(aminooxy) ethyl]carbamate (III, 28.0g, 0.159 mol; prepared as per the literature procedure depicted in Scheme 2) in dimethylformamide (56 ml) was added drop wise at 10°C under continuous stirring. The temperature of the reaction mass was allowed to warm to 25°C and then HOBt (21.5g, 0.159 mol) was added in small portions over a period of 15 minutes and the resulting mixture was further stirred at room temperature for 16 hours. The reaction was continuously monitored using thin layer chromatography using mixture of acetone and hexane (35 :65) as solvent system. After completion of reaction, the resulting mixture was filtered and the residue was washed with water (130 ml). The obtained white residue was suspended in water (130 ml) and the mixture stirred at 50°C for 3 hours. The resulting suspension was filtered, the residue dried under reduced pressure to obtain 51 g of (2S,5R)-N-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (IV) as off white solid in 73% yield.
Analysis:
Mass: 433.4 (M-l ); for Molecular Weight of 434.5 and Molecular Formula of C21H30N4O6;
1H-NMR (400MHz, CDC13): δ 9.32 (br s, 1H), 7.41 -7.26(m,5H), 5.41(br s, 1H), 5.06-4.88(dd, 2H), 3.98-3.96(d,lH), 3.91-3.90(m,2H), 3.39(m, 1H), 3.31-3.26(m, 2H), 3.04-3.01(d,lH), 2.77-2.74(d, 1H), 2.33-2.28(m, 1H), 2.03-1.93(m, 2H), 1.67-1.64(m, 1H), 1.44(s, 9H);
Purity as determined by HPLC: 99.4%.
Step 2: Preparation of (2S,5R)-iV-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (V):

A solution of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1] octane-2-carboxamide (IV, 38 g, 0.0875 mol) in a mixture of dimethylformamide and dichloromethane (2: 8, 76 ml: 304 ml), containing 10% Pd/C (7.6 g, 50% wet) was hydrogenated at 50 psi hydrogen atmosphere at 25°C for 3 hours. The resulting mixture was filtered through a celite pad. The residue was washed with dichloromethane (75 ml). The solvent from the combined filtrate was evaporated
under reduced pressure to obtain 30 g (25,5i?)-N-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide (V) as an oil, which was used as such for the next reaction without further purification.
Analysis:
Mass: 343.3 (M-l ) for Molecular Weight of 344.3 and Molecular Formula of C14H24N4O6.
Step 3: Preparation of (25,5R)-iV-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide,tetrabutyl ammonium salt (VI):

To a stirred solution of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide (V, 30.0 g, 0.0875 mol) in dimethylformamide (150 ml) was added sulphur trioxide dimethylformamide complex (16.06 g, 0.105 mol) in one portion, at 10°C. The reaction mass was stirred at the same temperature for 30 minutes and then allowed to warm to room temperature. After 2 hours, a solution of tetrabutylammonium acetate (31.6 g, 0.105 mol) in water (95 ml) was slowly added to the reaction mixture and stirred for another 2 hours. The solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2 x 60 ml) to obtain thick mass. This mass was partitioned between 1 : 1 mixture of dichloromethane (300 ml) and water (300 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (150 ml). The combined organic extracts were washed with water (3 x 150 ml) and dried over anhydrous sodium sulphate. The solvent was evaporated under reduced pressure and the resulting oily mass was triturated with ether (3 x 60 ml). Each time the ether layer was decanted and the residue was finally concentrated under reduced pressure to obtain the sticky mass. The so obtained material was purified by column chromatography over silica gel using mixture of methanol and dichloromethane as elution solvent. The solvent from the combined fractions was evaporated to obtain 47.5 g of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide,tetrabutyl ammonium salt as white foam in 70% yield.
Analysis:
Mass: 423.4 (M-l) as free sulphonic acid; for Molecular Weight of 665.9 and Molecular Formula of C30H59N5O9 S;
1H- NMR (400MHz, CDC13): δ 9.52(br s, 1H), 5.53(br s, 1H), 4.33(s, 1H), 3.95-3.92(m,3H), 3.37-3.27(m, 1 1H), 2.87-2.84(d, 1H), 2.35-2.30(m, 1H), 2.17(m, 1H), 1.96-1.88(m, 2H), 1.74-1.60(m,8 H), 1.47-1.40(m, 17H), 1.02-0.98(m, 12H).
Step 4: Preparation of (2S R)-iV-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (I):
Formula (I)

To a stirred solution of (2S,5i?)-N-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide, tetrabutyl ammonium salt (VI, 17 g, 0.0225 mol) in dichloromethane (85 ml) was added trifluoroacetic acid (85 ml) drop wise at -10°C over a period of 45 minutes. The resulting mass was further stirred at same temperature for 1 hour. The resulting reaction mixture was poured into cyclohexane (850 ml), stirred well for 30 minutes and the separated oily layer was collected. This procedure was repeated one more time and finally the separated oily layer was added to tert-butyl methyl ether (170 ml) under vigorous stirring at 25°C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with tert-butyl methyl ether (2 x 170 ml). The solid thus obtained was stirred with fresh dichloromethane (170 ml) for 30 minutes and filtered. The residual solid was dried at 45°C under reduced pressure to yield 7.3g of the titled compound in crude form. The obtained solid was further dissolved in water, (7.3 ml) and to this solution was added basic resin (Amberlyst A-26 -OH ion exchange resin, 4.4 g) under stirring. After 0.5 hour, the resin was filtered and to the filtrate isopropanol (51 ml) was added slowly at 25°C. The solution was further stirred for 12 hours. The separated solid was filtered and washed with additional isopropanol (7.5 ml) and dried under reduced pressure to obtain 4.3 g of (2S ,5R)-N-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide as off-white solid in 52 % yield.
Analysis:
Mass: 323.1 (M-l); for Molecular Weight of 324.31 and Molecular Formula of C9H16N4O7S; 1H-NMR (400MHz, D20): δ 4.07-4.06(d, 1H), 4.05-4.03(t, 2H), 3.96-3.94(d, 1H), 3.20(br s, 1H), 3.16-3.13(t, 2H), 3.02-2.99(d, 1H), 2.04-1.68(m, 4H);
Purity as determined by HPLC: 94.88%.

REF
| WO2015110969A3 * | Jan 21, 2015 | Nov 26, 2015 | Wockhardt Limited | Nitrogen containing compounds and their use as antibacterial agents |
| WO2015150941A1 * | Mar 12, 2015 | Oct 8, 2015 | Wockhardt Limited | A process for preparation of sodium (2s, 5r)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate |
| WO2016088863A1 * | Dec 4, 2015 | Jun 9, 2016 | Meiji Seikaファルマ株式会社 | Method for producing crystals of diazabicyclooctane derivative and stable lyophilized preparation |
| EP2931723A4 * | Dec 11, 2012 | Jun 1, 2016 | Fedora Pharmaceuticals Inc | New bicyclic compounds and their use as antibacterial agents and -lactamase inhibitors |
| US8933232 | Mar 29, 2013 | Jan 13, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| US8933233 | Mar 29, 2013 | Jan 13, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US8940897 | Mar 29, 2013 | Jan 27, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US8962843 | Mar 29, 2013 | Feb 24, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| US8962844 | Mar 29, 2013 | Feb 24, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US9120795 | Mar 14, 2014 | Sep 1, 2015 | Cubist Pharmaceuticals, Inc. | Crystalline form of a β-lactamase inhibitor |
| US9120796 | Oct 2, 2014 | Sep 1, 2015 | Cubist Pharmaceuticals, Inc. | B-lactamase inhibitor picoline salt |
| US9309245 | Apr 2, 2013 | Apr 12, 2016 | Entasis Therapeutics Limited | Beta-lactamase inhibitor compounds |
| US9393239 | Apr 15, 2014 | Jul 19, 2016 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and betalactamase inhibitors |
/////////////IN2015MU287, WO-2016120752, nacubactam, WOCKHARDT, NEW PATENT, WK ?, WK-?, WK?, CAS 1452458-86-4, C9 H16 N4 O7 S, 324.31, Beta lactamase inhibitor, Roche, Meiji Seika Pharma, Fedora Pharmaceuticals, nacubactam hydrate , PHASE 1, A diazabicyclooctane beta-lactamase inhibitor, bacterial infection, July 2016, phase 1 clinical development, RG-6080, 1452458-86-4, FPI-1459, OP-0595, Phase I , β-lactamase inhibitor, bacterial infections, Fedora parmaceuticals, Meiji Seika Pharma
NCCONC(=O)[C@@H]2CC[C@@H]1C[N@]2C(=O)N1OS(=O)(=O)O



 CH4O3S : 826
CH4O3S : 826








 The synthesis of the C27–C35 tetrahydrofuran fragment.
The synthesis of the C27–C35 tetrahydrofuran fragment. The synthesis of the C14–C21 aldehyde subfragment.
The synthesis of the C14–C21 aldehyde subfragment.






















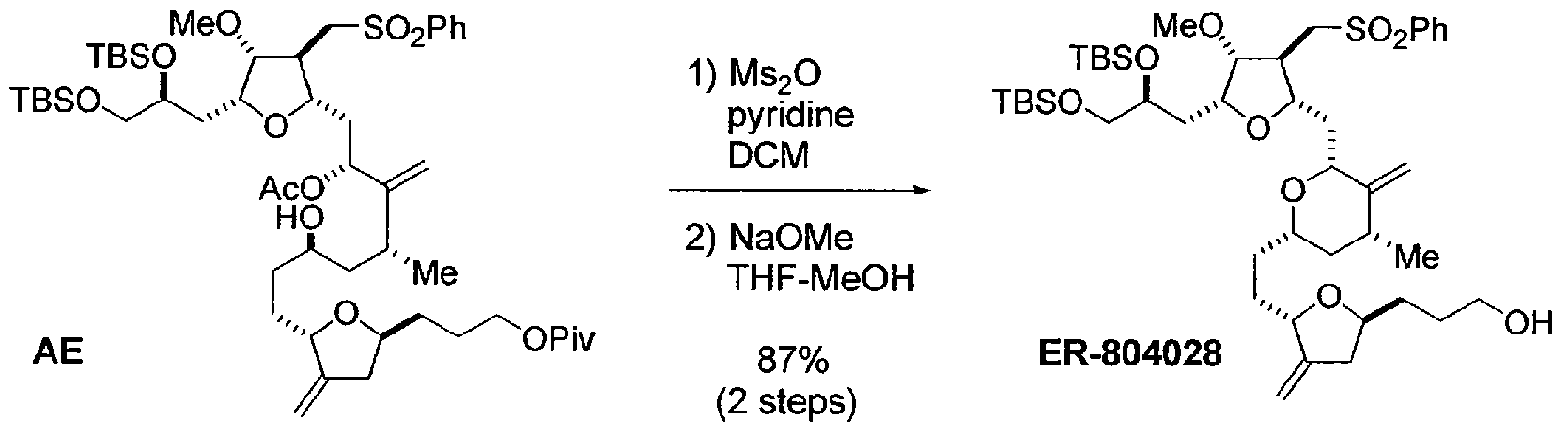
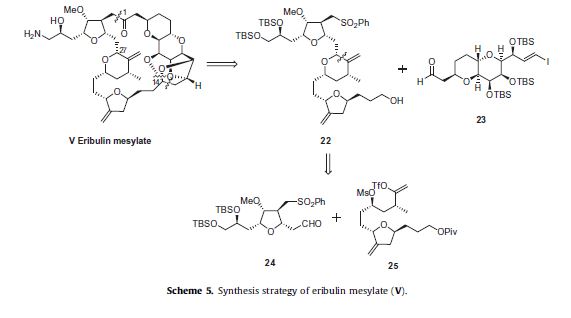
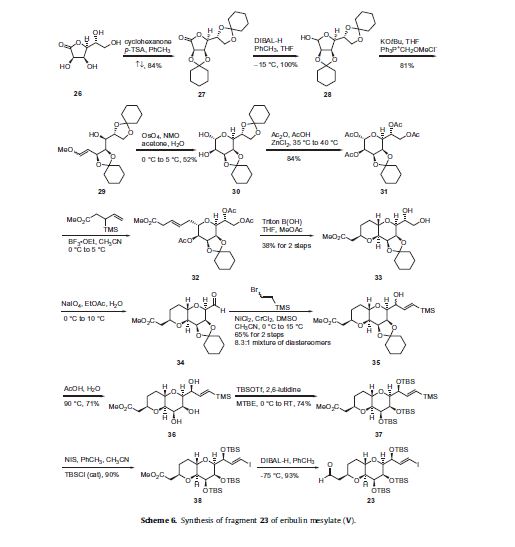
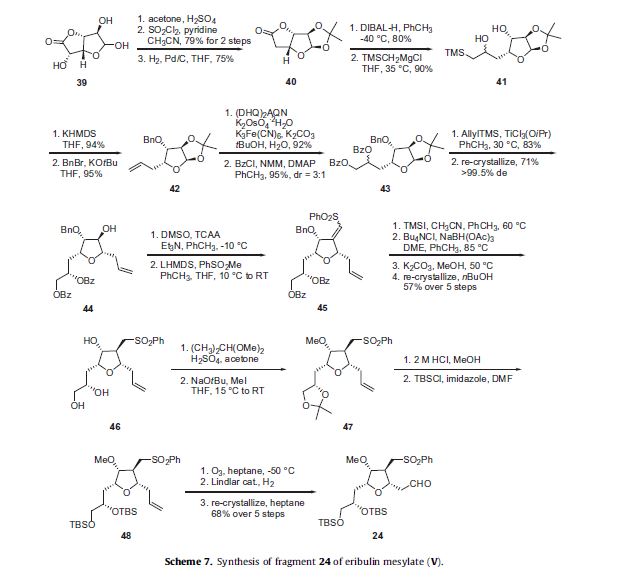
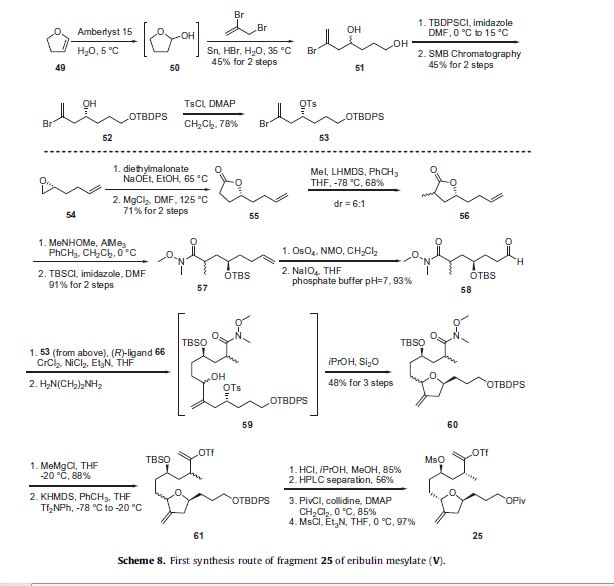
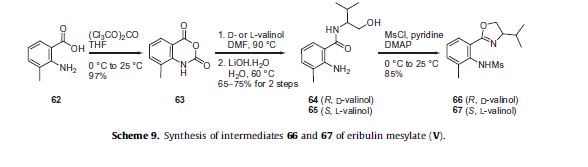
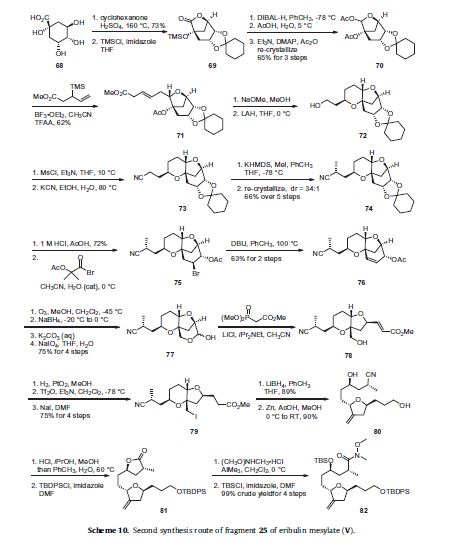
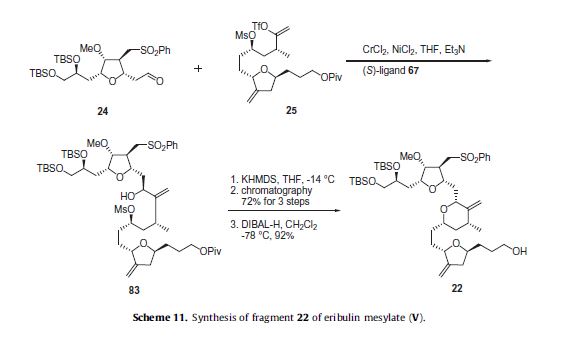
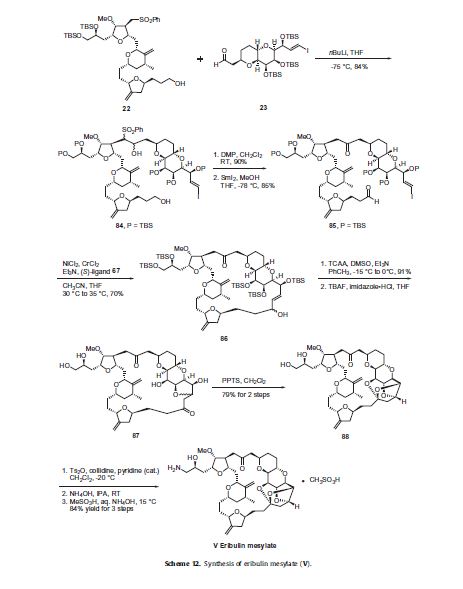
 Eribulin is a cut-down derivative of halichondrin B, which maintains most of its activity with significantly reduced complexity
Eribulin is a cut-down derivative of halichondrin B, which maintains most of its activity with significantly reduced complexity Synthesis of eribulin relies heavily on Nozaki–Hiyama–Kishi (NHK) coupling reactions to make key C–C bonds
Synthesis of eribulin relies heavily on Nozaki–Hiyama–Kishi (NHK) coupling reactions to make key C–C bonds
