
N-(5-chloropyridin-2-yl)-2-([4-(N,N-dimethylcarbamimidoyl)benzoyl]amino)-5-methoxybenzamide
Betrixaban:PRT-54021, PRT-021, MK-4448, PRT-054021
N- (5- chloro-2-pyridyl) -2 – [[4 – [(dimethylamino) methyl] benzoyl] amino] -5 – methoxy – benzamideCAS 330942-05-7
MW 451.91, C23H22ClN5O3
Venous Thromboembolism (VTE)
Millennium INNOVATOR
Takeda Pharmaceutical Co Ltd
Lee’s Pharmaceutical Holdings (Hong Kong) Ltd; Portola Pharmaceuticals Inc…DEVELOPERS
PHASE 3 for Venous Thromboembolism (VTE)

Portola Pharmaceuticals, under license from Takeda (formerly known as Millennium Pharmaceuticals), is developing betrixaban (was reported to be in phase III in November 2015), for treating venous thrombosis
In October 2015, betrixaban was granted Fast Track designation by the FDA for extended-duration prevention of VTE or blood clots in acute medically ill patients
Betrixaban (INN, codenamed PRT-054,021) is an anticoagulant drug which acts as a direct factor Xa inhibitor.[1] It is potent, orally active and highly selective for factor Xa, being selected from a group of similar compounds for its low hERG affinity.[2] Betrixaban has undergone human clinical trials for prevention of embolism after knee surgery,[3] and prevention of stroke following atrial fibrillation,[4] with promising results.[5] Betrixaban is currently being studied in a human clinical trial for extended duration thromboprophylaxis to prevent venous thromboembolism in acute medically ill patients.[6] Joint development with Portola was discontinued in 2011 by Merck.[7] Betrixaban is now being developed by Portola Pharmaceuticals.

Long-acting, oral, direct Factor Xa Inhibitor
Description
Betrixaban is an oral small molecule anticoagulant that directly inhibits the activity of Factor Xa, an important validated target in the blood coagulation pathway.Key Characteristics
Betrixaban has been specifically designed for chronic, once-a-day treatment. It has a half-life that supports true, once-daily dosing and a low peak-to-trough drug concentration ratio that minimizes anticoagulant variability. Betrixaban is primarily eliminated unchanged in the bile and has been studied in patients with all degrees of renal function, including those with severe renal impairment (excluding dialysis patients). Betrixaban is minimally metabolized through the Cytochrome 450 enzyme system, which may result in low potential for CYP-related drug interactions. Betrixaban is reversible with PRT4445, a universal Factor Xa inhibitor antidote that Portola is developing as a companion product.Potential Indications
Treatment or prevention of life-threatening blood clots (venous thromboembolism; VTE) in acute medically ill patients.Clinical Development
ClinicalTrials.gov Identifier:
NCT01583218
COMPLETION-August 2014
http://clinicaltrials.gov/ct2/show/NCT01583218
Portola has initiated its pivotal Phase 3 APEX Study to demonstrate the safety and efficacy of betrixaban for extended duration venous thromboembolism (VTE) prophylaxis for up to 35 days in acute medically ill patients with restricted mobility and certain risk factors. This randomized, double-blind, active-controlled, multicenter, multinational study will compare a once-daily dose of 80 mg of betrixaban for a total of 35 days (including both in the hospital and after discharge) with in-hospital administration of 40 mg of enoxaparin once daily for 6 to 14 days followed by placebo. The global study is expected to enroll approximately 6,850 patients at more than 400 study sites throughout the world. The primary objective of the trial is to demonstrate the superiority of betrixaban as compared to the current standard of care in the reduction of VTE-related events at 35 days while maintaining a favorable benefit to risk profile.
The APEX study is adequately powered to show a clinically relevant benefit with a p-value of less than 0.01 on the primary endpoint of total asymptomatic proximal DVT (as detected by ultrasound), symptomatic DVT (proximal or distal), non-fatal pulmonary embolism and VTE-related death. The first patient was enrolled in March 2012.
The safety and tolerability of betrixaban for stroke prevention was evaluated in 508 patients with atrial fibrillation in the Phase 2 EXPLORE-Xa dose-ranging study. Results were presented during a late-breaking session at the American College of Cardiology’s 59th Annual Scientific Session in March 2010. The data showed that a once-daily dose of oral betrixaban, given to patients with non-valvular atrial fibrillation or atrial flutter and at least one risk factor for stroke, reduced the incidence of major and clinically relevant non-major bleeds compared to dose-adjusted warfarin. In August 2010, additional pharmacodynamic data from a pre-specified analysis of EXPLORE-Xa showed a concentration dependent relationship and provided further evidence for the anticoagulant activity of betrixaban across all three doses studied in the clinical trial. The additional pharmacodynamic analysis provides information for dose selection for Phase 3 evaluation of betrixaban.
In 2007, positive top-line results from EXPERT were published in The Journal of Thrombosis and Haemostasis. This randomized, multi-center, Phase 2 in-hospital efficacy and safety study of the prevention of VTE compared betrixaban with enoxaparin in 215 patients undergoing knee replacement surgery.

Betrixaban (INN, codenamed PRT-054,021) is an anticoagulant drug which acts as a direct factor Xa inhibitor.[1] It is potent, orally active and highly selective for factor Xa, being selected from a group of similar compounds for its low hERG affinity.[2] Betrixaban has undergone human clinical trials for prevention of embolism after knee surgery,[3] and prevention of stroke following atrial fibrillation,[4] with promising results.[5]
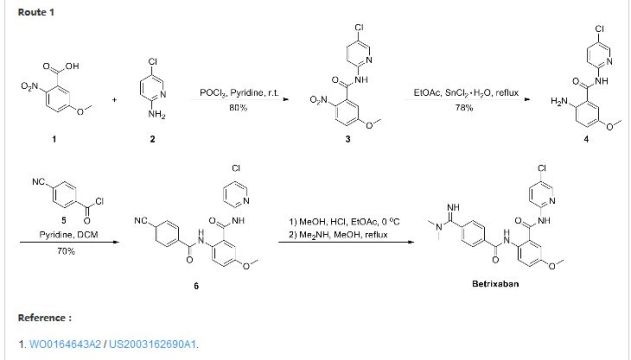

Patent Document CN1391555A first discloses a preparation method (see Scheme 1):

CN101595092A (See Scheme 2).

Patent Document CN102762538A (see Scheme 3).
[0013]

CN104693114
Machine translated from chinese please bear with names
http://www.google.com/patents/CN104693114A?cl=en

Preparation Example 1 shell song in Spanish

Under stirring, temperature 15 ~ 20 ° C, the compound of formula II 50. 0g (0 123mol, leq.) Was mixed with 500ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition continued at 25 The reaction was stirred for ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 700ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water; the filter cake was mixed by stirring with 500ml of acetone, the pH adjusted with triethylamine to 7-8; filtered; the cake at 40 ~ 45 ° C and dried under reduced pressure to give Pui Spanish song 45. 5g. . Yield: 82 0%; HPLC purity: 98.9%, of which 0.05% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Take the above Tony Qu Spanish 45.0g, at about 70 ° C under stirring dissolved in N, N- dimethylacetamide 180ml, a toluene solution of 360ml; cooling crystallization, filtration, the filter cake washed with an appropriate amount of acetone at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish HPLC purity 99.7%.
(+) LC-MS: m / z = 452 ([M + H] +). Che NMR (400MHz, DMS0-d6) S:…. 2 96 (s, 6H), 3 83 (s, 3H), 7. 06-7 09 (dd, 1H), 7. 55-7 59 ( m, 3H), 7. 80-7. 83 (dd, 1H), 8. 21-8. 23 (d, 1H), 8. 27-8. 30 (d, 2H), 8. 37-8. 40 (d, 1H), 8. 41-8. 43 (d, 1H), 10. 54 (br., 2H).
Preparation Example 2 Tony Qu Spanish maleate
Under stirring, temperature 0 ~ 5 ° C, dropping 2mol isopropyl magnesium chloride in tetrahydrofuran / L (available commercial available) 105ml (0 • 21mol, 8. 4eq) twenty methylamine hydrochloride 8. 91g (0 • llmol, 4. 4eq) in tetrahydrofuran 60ml of the suspension, the reaction solution obtained dimethylamine.
Under stirring, temperature 0 ~ 5 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, and then dropping the above reaction liquid dimethylamine; After the addition continued 10 The reaction was stirred for ~ 15 ° c, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 10 ~ 15 ° C, the reaction solution was added to an aqueous solution of 45g and 100ml dubbed maleic acid solution; the organic solvent was evaporated under reduced pressure and concentrated, filtered concentrate precipitated solid cake was washed with the right amount of water washing. Cake at 40 ~ 45 ° C and dried under reduced pressure to give Tony Qu Spanish maleate 12.lg. . Yield: 85 4%; HPLC purity: 98.6%, which is 0.03% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Take the above shellfish Spanish song maleate 10. 0g, at about 70 ° C under stirring dissolved in a mixed solvent of ethanol 50ml and 25ml of water, dropping water 150ml; cooling crystallization, filtration, the filter cake at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish maleate HPLC purity 99.9%.
: HNMR (400MHz, DMS〇-d6) 8: 3. 25 (s, 3H), 3. 32 (s, 3H), 3. 87 (s, 3H), 6. 02 (s, 2H) , 7. 19-7. 21 (dd, 1H), 7. 44-7. 45 (1H), 7. 75-7. 77 (d, 2H), 7. 97-9. 98 (d, 2H) , 8. 08-8. 13 (m, 3H), 8. 44-8. 45 (d, 1H), 9. 01 (br., 1H), 9. 37 (br., 1H), 11.04 (s , 1H), 11. 13 (s, 1H).
Preparation Example 3 Tony Spanish song of [0075] Example
Under stirring, temperature 25 ~ 30 ° C, isopropylmagnesium chloride in tetrahydrofuran was added dropwise a solution of 2mol / L (available commercially available) 81ml (0 • 161mol, 7eq) to 2mol / L dimethylamine THF Solution (commercially available can) 121ml (0 • 242mol, 10. 5eq) to give dimethylamine reaction solution.
Under stirring, temperature 25 ~ 30 ° C, the hydrochloride salt of a compound of formula II 10. 0g (0 023mol, leq.) Was mixed with 100ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition was complete The reaction continued stirring at 25 ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 210ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water; the filter cake with 90ml acetone was stirred and mixed, the pH adjusted with triethylamine to 7-8; filtration; cake was 45 ~ 50 ° C and dried under reduced pressure to give Pui Qu Spanish 8. 35g. Yield: 80.5%. HPLC purity: 98.7%, which is 0.03% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Preparation Example 4 shellfish Spanish song hydrochloride
Under stirring, temperature 15 ~ 20 ° C, dropping lmol / n-amyl magnesium bromide tetrahydrofuran solution (which can be commercialized available) 75ml (0 • 075mol, 3eq) to 2mol / L of dimethyl L amine in tetrahydrofuran (commercially available can) 56ml (0 • 113mol, 4. 5eq) to give dimethylamine reaction solution.
Under stirring, temperature 15 ~ 20 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition continued at 25 The reaction was stirred for ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 100ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water. Cake at 40 ~ 45 ° C and dried under reduced pressure to give Tony Qu Spanish hydrochloride 10.lg, yield:. 82 9%; HPLC purity: 99.0%, which is 0.02% dechlorination impurity VIII, from A impurities IX was not detected.
Take the above shellfish Spanish song hydrochloride 10. 0g, at about 70 ° C under stirring dissolved in N, N- dimethylacetamide 40ml, a toluene solution of 80ml; cooling crystallization, filtration, cake at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish hydrochloride HPLC purity 99.8%.
Preparation 5 shellfish Spanish song of [0082] Example
Under stirring, temperature 0 ~ 5 ° C, dropping lmol / diethyl zinc toluene solution of L (available commercially oriented) 50ml (0. 050mol, 2eq) to 2mol / L dimethylamine tetrahydrofuran (commercially available can) 28ml (0. 055mol, 2. 2eq) to give dimethylamine reaction solution.
Under stirring, temperature 0 ~ 5 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, and then dropping the above reaction liquid dimethylamine; After dropping 5 continues The reaction was stirred for ~ 10 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, in the next 5 ~ 10 ° C, the reaction mixture was added to about 2mol L dilute hydrochloric acid solution 70ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake was washed successively with a suitable amount of water; the filter cake with acetone l〇〇ml mixing, the pH adjusted with triethylamine to 7-8; filtered; the cake at 40 ~ 45 ° C under reduced pressed and dried to give Tony Qu Spanish 9. 03g. . Yield: 80 1%; HPLC purity: 99.0%, which is 0.02% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Preparation of compounds of Formula II Preparation Example 1
Methoxy-2-nitro – (5-chloro-pyridin-2-yl) -5 – benzamide (compound V) Preparation of [0086] (1) N-

(2) 2-Amino -N- (5- chloro – pyridin-2-yl) -5-methoxy – benzamide (compound IV) is prepared

(3) N- (5- chloro – pyridin-2-yl) -2- (4-cyano – benzoyl – amino) -5-methoxy – benzamide (compound II) is prepared

(+) LC-MS: m / z = 407 ([M + H] +). Insect NMR (400MHz, DMS0-d6) S:… 3 85 (s, 3H), 7 16-7 .19 (dd, 1H), 7. 39-7 41 (d, 1H), 7. 93- 7. 96 (d, 2H), 8. 02-8. 04 (m, 4H), 8. 13-8. 14 (d, 2H), 8. 42-8. 43 (d, 1H), 11. 06 (br. 2H).
Example 2 Preparation of the hydrochloride salt of the compound of formula II
at 10 ~ 20 ° C, a solution of a compound of formula IV 40. 0g (0 • 14mol, 1.Oeq) was dissolved in 400ml of tetrahydrofuran, a solution of cyanobenzoyl chloride (Compound III, can be commercialized available) 24 8g (0 15mol, 1.leq) and tetrahydrofuran solution 200ml dubbed, HPLC monitoring progress of the reaction;.. After the reaction was filtered, the filter cake washed with ethanol and after an appropriate amount, and dried under reduced pressure to obtain a compound of formula II hydrochloride . HPLC purity: 99.5%.
WO 2015176591
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015176591&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=FullTextExample 1: Preparation of Spanish Preparation and Form A half-L- malic acid shellfish song
At 55 ~ 60 ℃, the shellfish song Spanish 6.0g (13.3mmol), L- malic acid 1.1g (8.0mmol) was dissolved in tetrahydrofuran 70mL / water 7mL mixed solvent acetone was added with stirring 60mL, cooled to room temperature, Crystallization. Precipitated solid was filtered, and the resulting solid at 40 ~ 45 ℃ vacuum dried to give half L- malic acid shellfish Spanish song.
1H NMR(400MHz,MeOD)δ:2.355-2.419(dd,0.5H),2.735-2.781(dd,0.5H),3.226(s,6H),3.907(s,3H),4.302-4.326(dd,0.5H),7.195-7.224(dd,1H),7.448-7.455(d,1H),7.744-7.764(d,2H),7.821-7.849(dd,1H),8.145-8.165(d,2H),8.196-8.219(d,1H),8.238-8.261(d,1H),8.323-8.329(d,1H)。
Above 1 H-NMR results, δ: 3.907 (s, 3H) attributed to shellfish Spanish song molecule methyl CH 3 , 4.302-4.326 (dd, 0.5H) attributed to L- malic acid molecule methine CH , you can determine the song title product in shellfish Spanish and L- malic acid molar ratio of 2: 1.
PATENT
http://www.google.com.na/patents/EP2101760A2?cl=en
Example 2Preparation of the compound of Formula II

a. Gram scale preparation A slurry of the compound of Formula F (455 g, 1.0 eq.) in THF (4.67 kg,
10.3 parts) was prepared and adjusted to <10 0C. Lithium dimethyl amide was prepared as follows :hexyllithium (2.3 N/hexane, 2.45 L, 5.5 eq.) was added to dimethylamine solution (2 N/THF, 2.8 L, 5.5 eq.) maintaining <10 0C. The lithium dimethyl amide solution was charged into the slurry containing the compound of Formula F keeping the pot temperature of <10 0C. The reaction progress was monitored by in-process HPLC which confirmed that the amount of Formula F was <1.0 A%. A buffer solution of NaHCO3 (490 g, 1.1 parts, 5.7 eq.) and Na2CO3 (490 g, 1.1 parts, 4.5 eq.) in deionized water (6.6 kg, 14.51 parts) was prepared, and the above reaction mixture was transferred to this aqueous solution maintaining < 5 0C. The product precipitated out and the resulting slurry was adjusted to 20 0C over a period of 12 hr. The solid was filtered, and the resulting wet cake was washed with 3.5 kg (7.7 parts) of deionized water. The solid was filtered off using a coarse frit glass bench filter, and rinsed forwarded with cold (0-5 0C) absolute ethanol (628 g, 1.4 parts). The product was dried at 30-35 0C. Dry product was obtained in 458 g (73% yield). b. Kilogram scale preparation A slurry of the compound of Formula F (31.5 kg, 1.0 eq.) in THF (251 kg,8.0 parts) was prepared in a 780 L Hastelloy reactor (Reactor A) and adjusted to 0 0C (-3 to 3 0C). 2 M Dimethylamine in THF (161.0 kg, 5.0 eq.) and THF (63 kg, 2 parts) were charged into a 1900 L GLMS reactor (Reactor B) and adjusted to 0 0C (-3 to 3 0C) with maximum agitation. Hexyllithium (2.3 M, 97.2 kg, 4.5 eq.) was slowly charged to Reactor B while maintaining a max temperature of 10 0C. The pump and lines were rinsed forward to Reactor B with THF (3.2 kg). The Reactor B contents were adjusted to 0 0C (-3 to 3 0C), then transferred to Reactor A while keeping Reactor A temperature < 10 0C. The Reactor B pump and lines were rinsed forward with THF (31.4 kg, 1.0 part). The Reactor A contents were adjusted to 0 0C (-3 to 3 0C), and agitated at this temperature until the reaction was complete as verified by HPLC (1-2 hrs). After about 1 hr of agitation, in-process HPLC analysis indicated that 0 A% starting material remained (in-process criteria: max 1 A%). Reactor A contents were adjusted to -5 0C (-8 to -3 0C). In-process cleaning of Reactor B with water was performed. Two previously prepared aqueous solutions (NaHCO3 (35.0 kg, 1.1 parts) in water (236 kg, 7.5 parts), and Na2CO3 (35.0 kg 1.1 parts) in water (236 kg, 7.5 parts))were charged to Reactor B and adjusted to -3 0C (0 to 6 0C). Reactor A contents were transferred to Reactor B through an insulated line, maintaining the temperature of Reactor B at -8 0C to a maximum of 5 0C. The Reactor A pump and lines were rinsed forward with cold [-5 0C (-8 to -3 0C)] THF (31.4 kg, 1.0 part). Reactor B contents were adjusted to 22 0C (19-25 0C) and agitated for ca. 3 hrs. Slurry formation was visually confirmed, and Reactor B contents were filtered onto a 30″ centrifuge fitted with a filter cloth. The Reactor B pump and lines were rinsed forward onto the 30″ centrifuge fitted with a filter cloth with drinking water (63 kg, 2 parts). The wet filter cake (66.5 kg) was transferred back to Reactor B and submitted to a slurry wash in drinking water (1005 kg, 32 parts) at 22 0C (19-25) 0C for ca. 1 hr. The product was filtered onto the 30″ centrifuge (after in-process cleaning and fitting with a filter cloth), and the Reactor B lines and pump were rinsed forward with drinking water (63 kg, 2 parts). The water rinse was sampled for test by TDS, which was found to be 0.46%. The Reactor B pump, lines and wet filter cake were further rinsed with cold [0 0C (-3 to 3 0C)] ethanol (44 kg, 1.39 parts). The wet filter cake was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 35 0C. In-process LOD was 0% after ca. 24 hrs of drying, and the product was discharged (24.8 kg) in 76.7% yield. HPLC showed 98 % purity, with dechlorinated impurity at 1.14 %. Example 3
Preparation of the compound of Formula F Step 1. Synthesis of 2-nitro-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (C)

5-Methoxy-2-nitrobenzoic acid (A) (25.0 kg, 1.0 eq.), 2-amino-5- chloropyridine (B) (16.3 kg, 1.0 eq.), and acetonitrile (87.5 kg, 3.5 parts) were charged to a 380 L GLMS reactor. The reaction mixture was adjusted to 22 0C (19-25 0C) and anhydrous pyridine (30.0 kg, 3.0 eq.) was added. The pump and lines were rinsed forward with acetonitrile (22.5 kg, 0.9 parts), and the reactor contents were adjusted to a temperature of 19-22 0C. Phosphorous oxychloride (23.3 kg, 1.20 eq.) was charged to the contents of the reactor via a metering pump, while maintaining a temperature of 25 0C (22-28 0C). The metering pump and lines were rinsed forward with acetonitrile (12.5 kg, 0.5 parts), while keeping the temperature at 25 0C (22-28 0C). The reaction mixture normally turned from a slurry to a clear solution after the addition of about 1/3 of the POCI3. At the end of the addition, it became turbid. After complete addition, the reaction mixture was agitated at 25 0C (22-28 0C) for ca. 1 hr, at which time HPLC analysis confirmed reaction completion. The solution was cooled to 15 0C (12-18 0C) and drinking water (156.3 kg, 6.25 parts) was charged slowly while keeping reaction temperature of between 12 and 30 0C. The reaction mixture was then adjusted to 22 0C (19-25 0C) and agitated for ca. 5 hrs until exotherm ceased. Formation of a slurry was visually confirmed and the contents of the reactor were filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were washed forward onto the pressure nutsche with two portions of drinking water (62.5 kg, 2.5 parts each). The filtrate had a pH value of 7. The product (41.8 kg) was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 50 0C. After ca. 12 hrs, in-process LOD analysis indicated a solvent content of 0.72%. The dry product (C) was discharged (34.4 kg) with 88.2% yield and 99.1 % purity by HPLC. Step 2. Synthesis of 2-amino-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (D)


(17.2 kg, 1.1 eq.) and THF (92 kg, 3.5 parts). Reactor contents were agitated at 22 0C (19- 25 0C) until all of the solids had dissolved. The resulting solution was transferred to a lower receiver and the reactor was rinsed forward with THF (26 kg, 1 part). Compound D (26.4 kg, 1 eq.), THF (396 kg, 15 parts) and pyridine (2.90 kg, 0.4 eq.) were charged to a clean reactor. The pump and lines were rinsed forward with THF (34 kg, 1.3 parts). Via a metering pump, the 4-cyanobenzoyl chloride/THF solution was charged to the reactor, keeping the temperature at < 30 0C and rinsing forward with THF (ca. 10 kg). The resulting yellow-colored slurry was agitated at 22 0C (19-25 0C) for ca 2 hrs. In-process HPLC taken after 2 hrs showed a compound of Formula D content of 0%, indicating completion of the reaction. The slurry was filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, lines and wet cake were rinsed with three portions of ethanol (ca. 15 kg each). The wet filter cake was discharged (65.4 kg) and transferred back to the reactor for slurry wash in ethanol (317 kg, 12 parts) at 22 0C (19-25 0C) for ca. 1 hr. The slurry was filtered onto the pressure nutsche and the reactor, pump, lines, and wet filter cake were rinsed with two portions of ethanol (ca. 15 kg each) and two portions of THF (ca. 15 kg each). The wet filter cake was dried under vacuum with a maximum temperature of warm glycol bath (to heat the dryer jacket) of 40 0C. After 14.5 hrs of drying, LOD was 0.75%. The dried material was milled (screen 0.125″) to give 31.8 kg of product, which was dried under vacuum for another 10.5 hrs. LOD after drying was 1.8%, and the product was discharged (31.5 kg) in 74.8% yield (expected 60-90%). HPLC showed 100 % purity.
PATENT
http://www.google.com/patents/WO2011084519A1?cl=enU.S. Patent No. 6,376,515 B2 discloses a class of benzamide based compounds as specific factor Xa inhibitors. In particular, U.S. Patent No. 6,376,515 B2 describes a compound identified as Example 206, which is also disclosed in U.S. Patent No. 6,835,739 B2 as Example 206 and herein identified as betrixaban, which has the chemical formula of Formula I:

Scheme 1

Example 1: Preparation of betrixaban
 [0113] Dimethylformamide (13L) and hydrochloride (18 mL) were charged
into a reactor. Compound B (1 kg) was added followed by Compound A
(0.88 kg).
[0113] Dimethylformamide (13L) and hydrochloride (18 mL) were charged
into a reactor. Compound B (1 kg) was added followed by Compound A
(0.88 kg).
Compound A is commercially available or, just as with Compound B may be prepared using the methods described in Examples 4 and 5. The reaction mixture was cooled between 0 °C and -10 °C. EDC (0.752 kg) was added while maintaining the temperature between -10 °C and 0 °C. The reaction mixture was stirred until the content of
Compound B is below 0.10% area by HPLC. The reaction mixture was stirred until betrixaban started to crystallize. Acetone (26 L) was then added during a period of at least 1 hr while the temperature was maintained at between -10 °C and 0 °C. The suspension was then stirred for additional 2 hrs at a temperature of between 0 °C and 10 °C. The suspension was filtered and washed with cold acetone to give a wet product betrixaban. Example 2: Preparation of a maleate salt of betrixaban
 [0114] The wet betrixaban obtained above was reacted with maleic acid
(0.52 x weight of maleic acid/weight of dry betrixaban) in ethanol
(22.4 x volume of
[0114] The wet betrixaban obtained above was reacted with maleic acid
(0.52 x weight of maleic acid/weight of dry betrixaban) in ethanol
(22.4 x volume of
liquid/weight of dry betrixaban (v/w)) and purified water (5.7 x v/w) to form a betrixaban maleate salt. The solution of the betrixaban maleate salt was filtered and concentrated under vacuum until a final volume of 5.7 x v/w. Water (2 x v/w) was then added and the mixture was back concentrated until the same volume. The procedure of adding water and distil until a final volume of 5.7 x v/w was carried out until the molar ratio between the content of ethanol and the content of betrixaban maleate salt in the mixture was lower than, or equal to, 6. Betrixaban maleate salt crystallized during the removal of ethanol. The suspension was cooled to a temperature between 19 °C and 25 °C and stirred for not less than 2 hours at this temperature range. Betrixaban maleate salt was isolated by filtration, washed with water and dried under vacuum at a maximum temperature of 40 °C until the content of water was lower than, or equal to, 0.5 % w/w by Karl-Fisher. The purity of the maleate salt was determined to be greater than 99 % by HPLC. The betrixaban maleate isolated was in a crystalline form A which was concluded based on IR, DSC and XRPD results obtained, see Figures 3-5, respectively. The major peaks of XRPD pattern of crystalline form A are also listed in Table 2. Table 2: Betrixaban Form A XRPD Peak °2-Theta (2Θ0)
 Example 3: Synthesis of 2-nitro-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (C)
Example 3: Synthesis of 2-nitro-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (C)
 D E C
D E C
[0115] 5-Methoxy-2-nitrobenzoic acid (D) (25.0 kg, 1.0 eq.), 2-amino-5- chloropyridine (E) (16.3 kg, 1.0 eq.), and acetonitrile (87.5 kg) were charged to a 380 L glass-lined reactor. The reaction mixture was adjusted to 22 °C (19-25 °C) and anhydrous pyridine (30.0 kg, 3.0 eq.) was added. The pump and lines were rinsed forward with acetonitrile (22.5 kg), and the reactor contents were adjusted to a temperature of 19-22 °C. Phosphorous oxychloride (23.3 kg, 1.20 eq.) was charged to the contents of the reactor via a metering pump, while maintaining a temperature of 25 °C (22-28 °C). The metering pump and lines were rinsed forward with acetonitrile (12.5 kg), while keeping the temperature at 25 °C (22-28 °C). The reaction mixture normally turned from a slurry to a clear solution after the addition of about 1/3 of the POCI3. At the end of the addition, it became turbid. After complete addition, the reaction mixture was agitated at 25 °C (22- 28 °C) for ca. 1 hr, at which time HPLC analysis confirmed reaction completion. The solution was cooled to 15 °C (12-18 °C) and water (156.3 kg) was charged slowly while keeping reaction temperature of between 12 and 30 °C. The reaction mixture was then adjusted to 22 °C (19-25 °C) and agitated for ca. 5 hrs until exotherm ceased. Formation of a slurry was visually confirmed and the contents of the reactor were filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were washed forward onto the pressure nutsche with two portions of water (62.5 kg). The filtrate had a pH value of 7. The product (41.8 kg) was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. After ca. 12 hrs, in-process LOD analysis indicated a solvent content of 0.72%. The dry product (C) was discharged (34.4 kg) with 88.2% yield and 99.1 % purity by HPLC.
Exam le 4. Synthesis of 2-amino-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide
 Process A
Process A
[0116] To a 780 L Hastelloy reactor, Compound C (33 kg, 1.0 eq.), 5%> platinum carbon (sulfided, 0.33 kg) and dichloromethane (578 kg) were charged. Agitation was started and reactor contents were adjusted to 22 °C (19-25 °C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 28 °C (25-31 °C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 28 °C (25 to 31 °C; maximum 31 °C) until the reaction was complete by HPLC. After 16.5 hrs, the reaction was deemed complete after confirming the disappearance of starting material (0.472 A%). The contents of the reactor were circulated through a conditioned Celite™ (diatomaceous earth; Celite Co., Santa Barbara, Ca.) pad (0.2-0.5 kg Celite™ conditioned with 20-55 kg dichloromethane) prepared in a 8″ sparkler filter to remove the platinum catalyst. The reactor and Celite™ bed were rinsed forward with two portions of dichloromethane (83 kg). The filtrate was transferred to and concentrated in a 570 L glass-lined reactor under an atmospheric pressure to ca. 132 L. Ethanol (69 kg) was charged and concentration continued under atmospheric pressure to ca. 99 L. In-process NMR indicated that the dichloromethane content was 39%. Ethanol (69 kg) was charged again and concentration continued again to ca. 99 L. In-process NMR indicated that the dichloromethane content was 5%. The reaction mixture was then adjusted to 3 °C (0 to 6 °C), agitated for ca. 1 hr, and the resulting slurry filtered onto a jacketed pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were rinsed forward with cold [3 °C (0-6 °C)] ethanol (26 kg. The wet filter cake (36.6 kg) was dried under vacuum at 40-50 °C with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The dry product (B) was discharged (26.4 kg) in 89.5% yield. HPLC showed 98.4 A% purity, with dechlorinated impurity at 0.083 %.
Process B
[0117] To a 780 L Hastelloy reactor, Compound C (33 kg, 1.0 eq.), 5%> platinum carbon (sulfided, 0.33 kg) and dichloromethane (578 kg) were charged. Agitation was started and reactor contents were adjusted to 22 °C (19-25 °C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 26 °C (21 to 31 °C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 26 °C (21 to 31 °C; maximum 31 °C) until the reaction was complete by HPLC. After 16.5 hrs, the reaction was deemed complete after confirming the disappearance of starting material (0.472 A%). The contents of the reactor were circulated through a conditioned Celite™ pad (0.2-0.5 kg Celite™ conditioned with 20-55 kg dichloromethane) prepared in a 8″ sparkler filter to remove the platinum catalyst. The reactor and Celite™ bed were rinsed forward with two portions of dichloromethane (83 kg). The filtrate was transferred to and concentrated in a 570 L glass-lined reactor under vacuum and a maximum temperature of 45 °C to ca. 132 L. Ethanol (69 kg) was charged and concentration continued under vacuum and a maximum temperature of 45 °C to ca. 132 L. In-process NMR indicated that the dichloromethane content was 39%. Ethanol (69 kg) was charged again and concentration continued again to ca. 132 L. In-process NMR indicated that the dichloromethane content was 5%. The reaction mixture was then adjusted to 3 °C (0 to 6 °C), agitated for ca. 1 hr, and the resulting slurry filtered onto a jacketed pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were rinsed forward with cold [3 °C (0-6 °C)] ethanol (26 kg. The wet filter cake (36.6 kg) was dried under vacuum at 40-50 °C with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The dry product (B) was discharged (26.4 kg) in 89.5% yield. HPLC showed 98.4 A% purity, with dechlorinated impurity at 0.083 %.
Example 5. Synthesis of 4-(N,N-dimethylcarbamimidoyl)benzoic acid (A)
Process A
 Step 1: Amidine Formation
Step 1: Amidine Formation
[0118] To a tetrahydrofuran solution of 2M dimethylamine, 2.3M hexane solution of hexyllithium was slowly added over a period of at least three (3) hours while maintaining the temperature at between -8°C and -12°C. This solution was added to the tetrahydrofuran solution of ethyl-4-cyanobenzoate (F) while maintaining the temperature between -8°C and -12°C. The completion of the reaction was confirmed by HPLC, and the solution temperature was adjusted to between -8°C and 3°C. The reaction mixture was slowly added to the cold solution of aqueous sodium bicarbonate solution and the desired ethyl-4-(N,N-dimethylcarbamimidoyl)benzoate (G) was extracted with ethyl acetate. The ethyl acetate layer was dried, filtered and evaporated under vacuum to afford ethyl-4-(N,N-dimethylcarbamimidoyl)benzoate (G) as a white solid.
Step 2: Hydrolysis of ester
[0119] To a THF solution of ethyl -4(N,N-dimethylcarbamimidoyl)benzoate (G) was added an aqueous solution of lithium hydroxide (2 eq.) and the reaction mixture was stirred for 6 hr. The completion of the reaction was confirmed by HPLC. To the reaction mixture was added water, followed by extraction with ethyl acetate. The aqueous layer was acidified with 6N HCI to pH between 3-4 at which point the desired 4-(N,N- dimethylcarbamimidoyl)benzoic acid precipitated as the white solid. The white solid isolated was washed with hexane to afford 4-(N,N-dimethylcarbamimidoyl)benzoic acid as an hydrochloride salt (A).
Process B:
 Step 1: Ester Formation
Step 1: Ester Formation
[0120] To a methanolic solution of 4-cyanobenzoic acid was added concentrated sulfuric acid and refluxed the reaction for at least 12 hours. The completion of the reaction was confirmed by HPLC. The solution was cooled and the solvent was evaporated. To the residue was added ethyl acetate followed by washing with 10 % sodium hydroxide solution. The ethyl acetate layer was dried, filtered and evaporated to give desired 4-methyl cyanobenzoate as a white solid.
Step 2: Dimethylamidine formation
[0121] A stream of HCI (gas) was bubbled through a 0 °C solution of 4-methyl cyanobenzoate (1 mmol) in 50 mL of ethanol until saturation. The mixture was stirred at room temperature overnight and evaporated to afford compound P. The resulting residue was treated with dimethylamine hydrochloride (0.15 eq.) in 20 mL ethanol at reflux temperature for 4 hours. The solvent was removed at reduced pressure and the residue was washed with hexane to afford desired product Q as a light yellow solid.
Step 3: Ester hydrolysis
[0122] To a THF solution of ethyl-4(N,N-dimethylcarbamimidoyl)benzoate (Q) was added an aqueous solution of lithium hydroxide (2 eq.) and the reaction mixture was stirred for 6 hours. The completion of the reaction was confirmed by HPLC. To the reaction mixture was added water, followed by extraction with ethyl acetate. The aqueous layer was acidified with 6N HC1 to pH between 3-4 at which point the desired 4- (N,N-dimethylcarbamimidoyl)benzoic acid precipitated as the white solid. The white solid isolated was washed with hexane to afford 4-(N,N-dimethylcarbamimidoyl)benzoic acid as an hydrochloride salt (A).
Example 6: Preparation of betrixaban, free base
 [0123] To 100 mL round bottom flask, was added compound B (2.0 g,
obtained as in Example 4), compound A (1.98 g, obtained as in example
5), 20 mL N,N- dimethylacetamide. The reaction mixture was stirred
briefly so as to dissolve most of the solid, then con. HC1 (36
microliters) was added. To this thin slurry add EDC HCl (1.8 g total,
Aldrich) in 3 portions, 0.6 g each, 20 min apart. The reaction mixture
was stirred for 1.5 hours for complete reaction. [0124] To this reaction
was added 2.3 g sodium carbonate solution in 10 mL water while the
batch was cooled with water bath to keep the batch temperature 22-30 °C.
Vigorous agitation was required to keep the batch well mixed. Then 10
mL water was added. The batch was stirred at 22-25 °C for 30 min. After a
slurry was formed, 20 mL more water was added. The batch was stirred at
22 °C for 1 hour. The batch was filtered and the wet cake was washed
with 3×5 mL water, then 5 mL acetone. The cake was dried on the funnel
by suction. The weight of the dry cake is 2.95 g -2.92 g which is the
crude betrixaban. To purify the crude betrixaban obtained, 1.0 g of the
crude solid was mixed with 4 mL Ν,Ν-dimethylacetamide and heated to 70
°C for 30 min. Then add 8 mL toluene was added and the mixture was
heated for 30 min, then cooled to 22 °C over 1 h, then cooled to 0 °C,
aged at 0 °C for 2 hours, filtered, washed with 2×1 mL toluene. The cake
was dried on the funnel by suction to obtain 0.88 g pure betrixaban
(I).
[0123] To 100 mL round bottom flask, was added compound B (2.0 g,
obtained as in Example 4), compound A (1.98 g, obtained as in example
5), 20 mL N,N- dimethylacetamide. The reaction mixture was stirred
briefly so as to dissolve most of the solid, then con. HC1 (36
microliters) was added. To this thin slurry add EDC HCl (1.8 g total,
Aldrich) in 3 portions, 0.6 g each, 20 min apart. The reaction mixture
was stirred for 1.5 hours for complete reaction. [0124] To this reaction
was added 2.3 g sodium carbonate solution in 10 mL water while the
batch was cooled with water bath to keep the batch temperature 22-30 °C.
Vigorous agitation was required to keep the batch well mixed. Then 10
mL water was added. The batch was stirred at 22-25 °C for 30 min. After a
slurry was formed, 20 mL more water was added. The batch was stirred at
22 °C for 1 hour. The batch was filtered and the wet cake was washed
with 3×5 mL water, then 5 mL acetone. The cake was dried on the funnel
by suction. The weight of the dry cake is 2.95 g -2.92 g which is the
crude betrixaban. To purify the crude betrixaban obtained, 1.0 g of the
crude solid was mixed with 4 mL Ν,Ν-dimethylacetamide and heated to 70
°C for 30 min. Then add 8 mL toluene was added and the mixture was
heated for 30 min, then cooled to 22 °C over 1 h, then cooled to 0 °C,
aged at 0 °C for 2 hours, filtered, washed with 2×1 mL toluene. The cake
was dried on the funnel by suction to obtain 0.88 g pure betrixaban
(I).

Compound A is commercially available or, just as with Compound B may be prepared using the methods described in Examples 4 and 5. The reaction mixture was cooled between 0 °C and -10 °C. EDC (0.752 kg) was added while maintaining the temperature between -10 °C and 0 °C. The reaction mixture was stirred until the content of
Compound B is below 0.10% area by HPLC. The reaction mixture was stirred until betrixaban started to crystallize. Acetone (26 L) was then added during a period of at least 1 hr while the temperature was maintained at between -10 °C and 0 °C. The suspension was then stirred for additional 2 hrs at a temperature of between 0 °C and 10 °C. The suspension was filtered and washed with cold acetone to give a wet product betrixaban. Example 2: Preparation of a maleate salt of betrixaban

liquid/weight of dry betrixaban (v/w)) and purified water (5.7 x v/w) to form a betrixaban maleate salt. The solution of the betrixaban maleate salt was filtered and concentrated under vacuum until a final volume of 5.7 x v/w. Water (2 x v/w) was then added and the mixture was back concentrated until the same volume. The procedure of adding water and distil until a final volume of 5.7 x v/w was carried out until the molar ratio between the content of ethanol and the content of betrixaban maleate salt in the mixture was lower than, or equal to, 6. Betrixaban maleate salt crystallized during the removal of ethanol. The suspension was cooled to a temperature between 19 °C and 25 °C and stirred for not less than 2 hours at this temperature range. Betrixaban maleate salt was isolated by filtration, washed with water and dried under vacuum at a maximum temperature of 40 °C until the content of water was lower than, or equal to, 0.5 % w/w by Karl-Fisher. The purity of the maleate salt was determined to be greater than 99 % by HPLC. The betrixaban maleate isolated was in a crystalline form A which was concluded based on IR, DSC and XRPD results obtained, see Figures 3-5, respectively. The major peaks of XRPD pattern of crystalline form A are also listed in Table 2. Table 2: Betrixaban Form A XRPD Peak °2-Theta (2Θ0)


[0115] 5-Methoxy-2-nitrobenzoic acid (D) (25.0 kg, 1.0 eq.), 2-amino-5- chloropyridine (E) (16.3 kg, 1.0 eq.), and acetonitrile (87.5 kg) were charged to a 380 L glass-lined reactor. The reaction mixture was adjusted to 22 °C (19-25 °C) and anhydrous pyridine (30.0 kg, 3.0 eq.) was added. The pump and lines were rinsed forward with acetonitrile (22.5 kg), and the reactor contents were adjusted to a temperature of 19-22 °C. Phosphorous oxychloride (23.3 kg, 1.20 eq.) was charged to the contents of the reactor via a metering pump, while maintaining a temperature of 25 °C (22-28 °C). The metering pump and lines were rinsed forward with acetonitrile (12.5 kg), while keeping the temperature at 25 °C (22-28 °C). The reaction mixture normally turned from a slurry to a clear solution after the addition of about 1/3 of the POCI3. At the end of the addition, it became turbid. After complete addition, the reaction mixture was agitated at 25 °C (22- 28 °C) for ca. 1 hr, at which time HPLC analysis confirmed reaction completion. The solution was cooled to 15 °C (12-18 °C) and water (156.3 kg) was charged slowly while keeping reaction temperature of between 12 and 30 °C. The reaction mixture was then adjusted to 22 °C (19-25 °C) and agitated for ca. 5 hrs until exotherm ceased. Formation of a slurry was visually confirmed and the contents of the reactor were filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were washed forward onto the pressure nutsche with two portions of water (62.5 kg). The filtrate had a pH value of 7. The product (41.8 kg) was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. After ca. 12 hrs, in-process LOD analysis indicated a solvent content of 0.72%. The dry product (C) was discharged (34.4 kg) with 88.2% yield and 99.1 % purity by HPLC.
Exam le 4. Synthesis of 2-amino-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide

[0116] To a 780 L Hastelloy reactor, Compound C (33 kg, 1.0 eq.), 5%> platinum carbon (sulfided, 0.33 kg) and dichloromethane (578 kg) were charged. Agitation was started and reactor contents were adjusted to 22 °C (19-25 °C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 28 °C (25-31 °C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 28 °C (25 to 31 °C; maximum 31 °C) until the reaction was complete by HPLC. After 16.5 hrs, the reaction was deemed complete after confirming the disappearance of starting material (0.472 A%). The contents of the reactor were circulated through a conditioned Celite™ (diatomaceous earth; Celite Co., Santa Barbara, Ca.) pad (0.2-0.5 kg Celite™ conditioned with 20-55 kg dichloromethane) prepared in a 8″ sparkler filter to remove the platinum catalyst. The reactor and Celite™ bed were rinsed forward with two portions of dichloromethane (83 kg). The filtrate was transferred to and concentrated in a 570 L glass-lined reactor under an atmospheric pressure to ca. 132 L. Ethanol (69 kg) was charged and concentration continued under atmospheric pressure to ca. 99 L. In-process NMR indicated that the dichloromethane content was 39%. Ethanol (69 kg) was charged again and concentration continued again to ca. 99 L. In-process NMR indicated that the dichloromethane content was 5%. The reaction mixture was then adjusted to 3 °C (0 to 6 °C), agitated for ca. 1 hr, and the resulting slurry filtered onto a jacketed pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were rinsed forward with cold [3 °C (0-6 °C)] ethanol (26 kg. The wet filter cake (36.6 kg) was dried under vacuum at 40-50 °C with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The dry product (B) was discharged (26.4 kg) in 89.5% yield. HPLC showed 98.4 A% purity, with dechlorinated impurity at 0.083 %.
Process B
[0117] To a 780 L Hastelloy reactor, Compound C (33 kg, 1.0 eq.), 5%> platinum carbon (sulfided, 0.33 kg) and dichloromethane (578 kg) were charged. Agitation was started and reactor contents were adjusted to 22 °C (19-25 °C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 26 °C (21 to 31 °C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 26 °C (21 to 31 °C; maximum 31 °C) until the reaction was complete by HPLC. After 16.5 hrs, the reaction was deemed complete after confirming the disappearance of starting material (0.472 A%). The contents of the reactor were circulated through a conditioned Celite™ pad (0.2-0.5 kg Celite™ conditioned with 20-55 kg dichloromethane) prepared in a 8″ sparkler filter to remove the platinum catalyst. The reactor and Celite™ bed were rinsed forward with two portions of dichloromethane (83 kg). The filtrate was transferred to and concentrated in a 570 L glass-lined reactor under vacuum and a maximum temperature of 45 °C to ca. 132 L. Ethanol (69 kg) was charged and concentration continued under vacuum and a maximum temperature of 45 °C to ca. 132 L. In-process NMR indicated that the dichloromethane content was 39%. Ethanol (69 kg) was charged again and concentration continued again to ca. 132 L. In-process NMR indicated that the dichloromethane content was 5%. The reaction mixture was then adjusted to 3 °C (0 to 6 °C), agitated for ca. 1 hr, and the resulting slurry filtered onto a jacketed pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were rinsed forward with cold [3 °C (0-6 °C)] ethanol (26 kg. The wet filter cake (36.6 kg) was dried under vacuum at 40-50 °C with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The dry product (B) was discharged (26.4 kg) in 89.5% yield. HPLC showed 98.4 A% purity, with dechlorinated impurity at 0.083 %.
Example 5. Synthesis of 4-(N,N-dimethylcarbamimidoyl)benzoic acid (A)
Process A

[0118] To a tetrahydrofuran solution of 2M dimethylamine, 2.3M hexane solution of hexyllithium was slowly added over a period of at least three (3) hours while maintaining the temperature at between -8°C and -12°C. This solution was added to the tetrahydrofuran solution of ethyl-4-cyanobenzoate (F) while maintaining the temperature between -8°C and -12°C. The completion of the reaction was confirmed by HPLC, and the solution temperature was adjusted to between -8°C and 3°C. The reaction mixture was slowly added to the cold solution of aqueous sodium bicarbonate solution and the desired ethyl-4-(N,N-dimethylcarbamimidoyl)benzoate (G) was extracted with ethyl acetate. The ethyl acetate layer was dried, filtered and evaporated under vacuum to afford ethyl-4-(N,N-dimethylcarbamimidoyl)benzoate (G) as a white solid.
Step 2: Hydrolysis of ester
[0119] To a THF solution of ethyl -4(N,N-dimethylcarbamimidoyl)benzoate (G) was added an aqueous solution of lithium hydroxide (2 eq.) and the reaction mixture was stirred for 6 hr. The completion of the reaction was confirmed by HPLC. To the reaction mixture was added water, followed by extraction with ethyl acetate. The aqueous layer was acidified with 6N HCI to pH between 3-4 at which point the desired 4-(N,N- dimethylcarbamimidoyl)benzoic acid precipitated as the white solid. The white solid isolated was washed with hexane to afford 4-(N,N-dimethylcarbamimidoyl)benzoic acid as an hydrochloride salt (A).
Process B:

[0120] To a methanolic solution of 4-cyanobenzoic acid was added concentrated sulfuric acid and refluxed the reaction for at least 12 hours. The completion of the reaction was confirmed by HPLC. The solution was cooled and the solvent was evaporated. To the residue was added ethyl acetate followed by washing with 10 % sodium hydroxide solution. The ethyl acetate layer was dried, filtered and evaporated to give desired 4-methyl cyanobenzoate as a white solid.
Step 2: Dimethylamidine formation
[0121] A stream of HCI (gas) was bubbled through a 0 °C solution of 4-methyl cyanobenzoate (1 mmol) in 50 mL of ethanol until saturation. The mixture was stirred at room temperature overnight and evaporated to afford compound P. The resulting residue was treated with dimethylamine hydrochloride (0.15 eq.) in 20 mL ethanol at reflux temperature for 4 hours. The solvent was removed at reduced pressure and the residue was washed with hexane to afford desired product Q as a light yellow solid.
Step 3: Ester hydrolysis
[0122] To a THF solution of ethyl-4(N,N-dimethylcarbamimidoyl)benzoate (Q) was added an aqueous solution of lithium hydroxide (2 eq.) and the reaction mixture was stirred for 6 hours. The completion of the reaction was confirmed by HPLC. To the reaction mixture was added water, followed by extraction with ethyl acetate. The aqueous layer was acidified with 6N HC1 to pH between 3-4 at which point the desired 4- (N,N-dimethylcarbamimidoyl)benzoic acid precipitated as the white solid. The white solid isolated was washed with hexane to afford 4-(N,N-dimethylcarbamimidoyl)benzoic acid as an hydrochloride salt (A).
Example 6: Preparation of betrixaban, free base

| WO2012031017A1 * | Aug 31, 2011 | Mar 8, 2012 | Merck Sharp & Dohme Corp. | CRYSTALLINE FORMS OF A FACTOR Xa INHIBITOR |
| WO2013033370A1 * | Aug 30, 2012 | Mar 7, 2013 | Portola Pharmaceuticals, Inc. | Prevention and treatment of thrombosis in medically ill patients |
| US8946269 | Aug 31, 2011 | Feb 3, 2015 | Portola Pharmaceuticals, Inc. | Crystalline forms of a factor Xa inhibitor |
| WO2004083174A2 * | Mar 17, 2004 | Sep 30, 2004 | Timur Gangor | Sulfonyl-amidino containing and tetrahydropyrimidino containing compounds as factor xa inhibitors |
| WO2008057972A1 | Nov 1, 2007 | May 15, 2008 | Millennium Pharm Inc | Methods of synthesizing pharmaceutical salts of a factor xa inhibitor |
| US6376515 | Feb 28, 2001 | Apr 23, 2002 | Cor Therapeutics, Inc. | Benzamides and related inhibitors of factor Xa |
| US6835739 | Oct 15, 2003 | Dec 28, 2004 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US6844367 | Sep 15, 2000 | Jan 18, 2005 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US61287680 |
References
- Eriksson BI, Quinlan DJ, Weitz JI (2009). “Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor xa inhibitors in development”. Clinical Pharmacokinetics48 (1): 1–22. PMID19071881.
- Zhang P, Huang W, Wang L, Bao L, Jia ZJ, Bauer SM, Goldman EA, Probst GD, Song Y, Su T, Fan J, Wu Y, Li W, Woolfrey J, Sinha U, Wong PW, Edwards ST, Arfsten AE, Clizbe LA, Kanter J, Pandey A, Park G, Hutchaleelaha A, Lambing JL, Hollenbach SJ, Scarborough RM, Zhu BY (April 2009). “Discovery of betrixaban (PRT054021), N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide, a highly potent, selective, and orally efficacious factor Xa inhibitor”. Bioorganic & Medicinal Chemistry Letters19 (8): 2179–85. doi:10.1016/j.bmcl.2009.02.111. PMID19297154.
- Turpie AG, Bauer KA, Davidson BL, Fisher WD, Gent M, Huo MH, Sinha U, Gretler DD (January 2009). “A randomized evaluation of betrixaban, an oral factor Xa inhibitor, for prevention of thromboembolic events after total knee replacement (EXPERT)”. Thrombosis and Haemostasis101 (1): 68–76. PMID19132191.
- Piccini, J. P.; Lopes, R. D.; Mahaffey, K. W. (2010). “Oral factor Xa inhibitors for the prevention of stroke in atrial fibrillation”. Current Opinion in Cardiology25 (4): 312. doi:10.1097/HCO.0b013e32833a524f. PMID20520539. edit
- Sobieraj-Teague, M.; O’donnell, M.; Eikelboom, J. (2009). “New Anticoagulants for Atrial Fibrillation”. Seminars in Thrombosis and Hemostasis35 (5): 515–24. doi:10.1055/s-0029-1234147. PMID19739042. edit
- 6 Cohen, Alexander T.; Harrington, Robert; Goldhaber, Samuel Z.; Hull, Russell; Gibson, C. Michael; Hernandez, Adrian F.; Kitt, Michael M.; Lorenz, Todd J. (2014-01-01). “The design and rationale for the Acute Medically Ill Venous Thromboembolism Prevention with Extended Duration Betrixaban (APEX) study”. American Heart Journal 167 (3). doi:10.1016/j.ahj.2013.11.006.
- 7
 |
|
| Systematic (IUPAC) name | |
|---|---|
N-(5-chloropyridin-2-yl)-2-([4-(N,N-dimethylcarbamimidoyl)benzoyl]amino)-5-methoxybenzamide
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Number | 330942-05-7 |
| ATC code | None |
| PubChem | CID: 10275777 |
| ChemSpider | 18981107 |
| UNII | 74RWP7W0J9 |
| ChEMBL | CHEMBL512351 |
| Chemical data | |
| Formula | C23H22ClN5O3 |
| Molecular mass | 451.905 g/mol |
/////////////CN(C)C(=N)C1=CC=C(C=C1)C(=O)NC2=C(C=C(C=C2)OC)C(=O)NC3=NC=C(C=C3)Cl
No comments:
Post a Comment