
CFI-402257
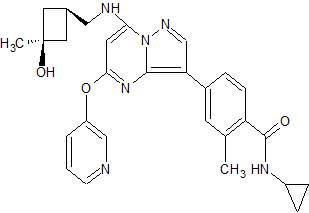
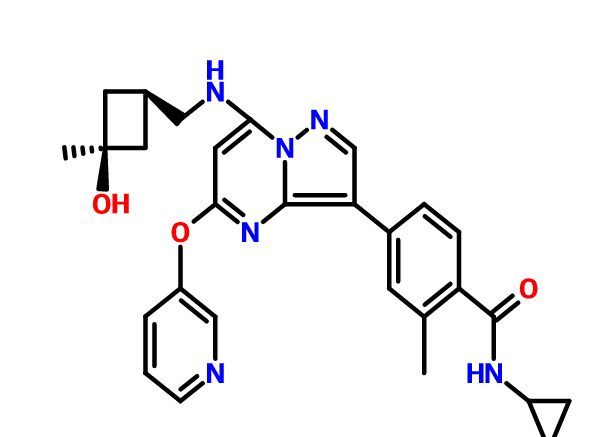
N-cyclopropyl-4-(7-((((1s,3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5-(pyridin-2-yloxy)pyrazolo[1,5-a]pyridin-3-yl)-2-methylbenzamide
N-cyclopropyl-4-(7-( (((Is, 3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5- (pyridin-3-yloxy)pyrazolol 1 , 5-a ]pyrimidin-3-yl)-2-methylbenzamide
CAS 1610759-22-2 (free base); 1610677-37-6 (HCl)
MF: C29H31N5O3
MW: 497.2427
MF: C29H31N5O3
MW: 497.2427
CFI-402257 is a highly potent and selective TTK (threonine tyrosine kinase) Inhibitor ((TTK Ki = 0.1 nM) with potential anticancer activity. TTK is an essential chromosomal regulator and is overexpressed in aneuploid tumors. High TTK levels correlate with a high tumor grade11 and poor patient outcomes. TTK inhibition are associated with a disabled mitotic checkpoint, resulting in chromosome segregation errors, aneuploidy, and cell death.
Synthesis
SYN OF INTERMEDIATE
SYNTHESIS COLOUR INDICATED
SYN OF INTERMEDIATES
IF YOU HAVE ENJOYED IT .........EMAIL ME amcrasto@gmail.com, +919323115463, India

INDIA FLAG

Protein kinases have been the subject of extensive study in the search for new therapeutic agents in various diseases, for example, cancer. Protein kinases are known to mediate intracellular signal transduction by effecting a phosphoryl transfer from a nucleoside triphosphate to a protein acceptor that is involved in a signaling pathway. There are a number of kinases and pathways through which extracellular and other stimuli cause a variety of cellular responses to occur inside the cell.
Human TTK protein kinase (TTK), also known as tyrosine threonine kinase, dual specificity protein kinase TTK, Monopolar Spindle 1 (Mpsl) and Phosphotyrosine -Picked Threonine Kinase (PYT), is a conserved multispecific kinase that is capable of phosphorylating serine, threonine and tyrosine residues when expressed in E. coli (Mills et al., J. Biol. Chem. 22(5): 16000-16006 (1992)). TTK mRNA is not expressed in the majority of physiologically normal tissues in human (Id). TTK mRNA is expressed in some rapidly proliferating tissues, such as testis and thymus, as well as in some tumors (for example, TTK mRNA was not expressed in renal cell carcinoma, was expressed in 50% of breast cancer samples, was expressed in testicular tumors and ovarian cancer samples) (Id). TTK is expressed in some cancer cell lines and tumors relative to normal counterparts (Id.; see also WO 02/068444 Al).
Therefore, agents which inhibit a protein kinase, in particular TTK, have the potential to treat cancer. There is a need for additional agents which can act as protein kinase inhibitors, in particular TTK inhibitors.
In addition, cancer recurrence, drug resistance or metastasis is one of the major challenges in cancer therapies. Cancer patients who responded favorably to the initial anticancer therapy often develop drug resistance and secondary tumors that lead to the relapse of the disease. Recent research evidences suggest that the capability of a tumor to grow and propagate is dependent on a small subset of cells within the tumor. These cells are termed tumor-initiating cells (TICs) or cancer stem cells. It is thought that the TICs are responsible for drug resistance, cancer relapse and metastasis. Compounds that can inhibit the growth and survival of these tumor-initiating cells can be used to treat cancer, metastasis or prevent recurrence of cancer. Therefore, a need exists for new compounds that can inhibit the growth and survival of tumor- imitating cells.
PATENT
WO 2015070349
A4: N-cyclopropyl-4-(7-( (((Is, 3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5- (pyridin-3-yloxy)pyrazolol 1 , 5-a ]pyrimidin-3-yl)-2-methylbenzamide hydrochloride and its free base
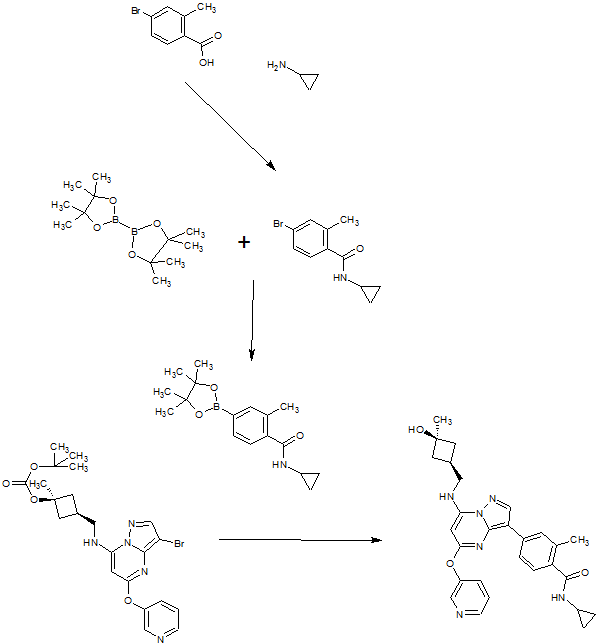
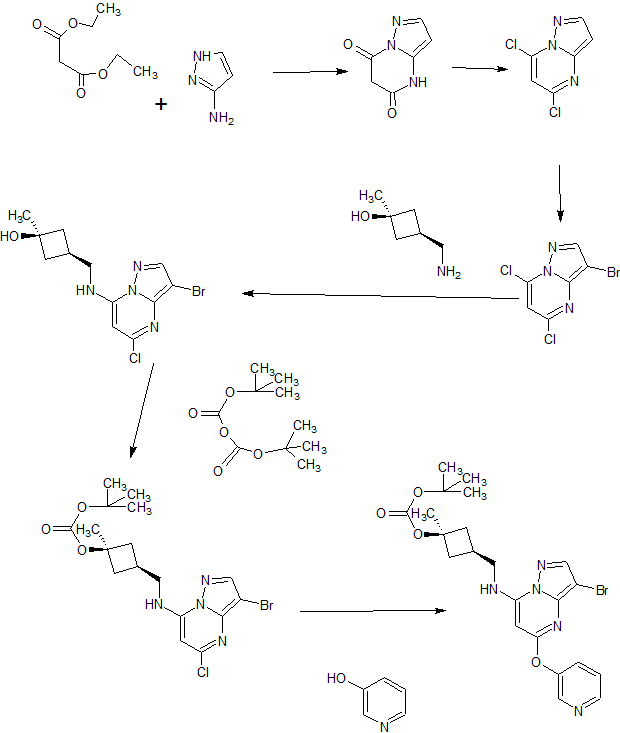
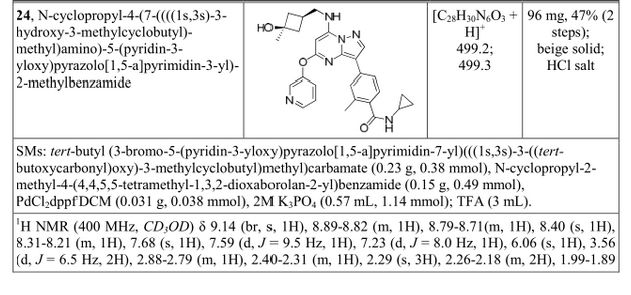
A). Through Boc deprotection: A mixture of tert-butyl (3- bromo-5-(pyridin-3-yloxy)pyrazolo[l,5-a]pyrimidin-7- yl)(((ls,3s)-3-((tert-butoxycarbonyl)oxy)-3- methylcyclobutyl)methyl)carbamate (0.23 g, 0.38 mmol), N- cyclopropyl-2-methyl-4-(4,4,5,5-tetramethyl-l,3,2- 
dioxaborolan-2-yl)benzamide (0.15 g, 0.49 mmol), PdC dppfDCM (0.15 g, 0.49 mmol), and 2M K3P04 (0.57 mL, 1.14 mmol) in THF (4 mL) was charged with Ar and heated in the microwave at 130 °C for 3 h. Water and EtOAc were added to separate the phases and the aqueous phase was extracted with EtOAc. The combined organic extracts were dried over NaSC>4, filtered and concentrated. The crude product was purified by flash chromatography (gradient: EtOAc/hex 20-60%) to give a yellow oil.
The above intermediate was dissolved in DCM (10 mL) and treated with TFA (3 mL) at rt for 3 h. After reaction completion, solvent was removed in vacuo and the crude product was dissolved in MeOH (5 mL). The mixture was filtered and purified by prep-HPLC. The compound was passed through a PoraPak cartridge and triturated with Et20 to give the title compound as a free base (white solid). The free base was dissolved in MeOH (5 mL), and HC1 (1 M Et20, 2 equiv) was then added slowly. Solvent was removed in vacuo to give the title compound as a beige solid in HC1 salt (96 mg, 47% over 2 steps). ¾ NMR (400 MHz, CD3OD) δ ppm 9.14 (br. s, 1H), 8.89-8.82 (m, 1H), 8.79-8.71 (m, 1H), 8.40 (s, 1H), 8.31-8.21 (m, 1H), 7.68 (s, 1H), 7.59 (d, J = 9.5 Hz, 1H), 7.23 (d, J= 8.0 Hz, 1H), 6.06 (s, 1H), 3.56 (d, J= 6.5 Hz, 2H), 2.88-2.79 (m, 1H),
2.40-2.31 (m, 1H), 2.29 (s, 3H), 2.26-2.18 (m, 2H), 1.99-1.89 (m, 2H), 1.37 (s, 3H),
0.85-0.76 (m, 2H), 0.63-0.53 (m, 2H); MS ESI [M + H]+ 499.3, calcd for [C^HsoNeOs +
H]+ 499.2. HPLC purity: 99.5% at 254 nm.
B). Through PMB deprotection: A mixture of N- cyclopropyl-4-(7-((((ls,3s)-3-hydroxy-3- methylcyclobutyl)methyl)(4-methoxybenzyl)amino)-5- (pyridin-3-yloxy)pyrazolo[l,5-a]pyrimidin-3-yl)-2- methylbenzamide (9.6 g, 15.5 mmol), TFA (50 mL) in DCE
(70 mL) was heated in an oil bath at 50 °C for 4 h. After reaction completion, solvent was removed in vacuo and the crude product was dissolved in a mixture of MeOH/DCM (100 mL/25 mL). 2M Na2CC (150 mL) was then added and the resulting mixture was stirred at rt for 30 min. The reaction mixture was diluted with DCM and the phases were separated. The aqueous phase was extracted with DCM and the combined organic extracts were washed with water, dried over MgSC , filtered and concentrated. The crude product was triturated and sonicated in a mixture of DCM/Et20 (10 mL/70 mL) to give the title compound as a off white solid in free base (5.9 g, 77%). Ti NMR (400 MHz, CD3OD) δ ppm 8.58-8.53 (m, 1H), 8.50-8.46 (m, 1H), 8.36 (s, 1H), 7.86-7.80 (m, 1H), 7.76-7.72 (m, 1H), 7.61-7.55 (m, 2H), 7.18 (d, J = 8.0 Hz, 1H), 5.92 (s, 1H), 3.52 (d, J = 6.8 Hz, 2H), 2.86-2.77 (m, 1H), 2.38-2.28 (m, 1H), 2.25 (s, 3H), 2.24-2.18 (m, 2H), 1.99-1.88 (m, 2H), 1.37 (s, 3H), 0.84-0.75 (m, 2H), 0.64-0.54 (m, 2H); MS ESI [M + H]+ 499.2, calcd for [CzsHsoNgOs + H]+ 499.2. HPLC purity: 96.1% at 235 nm.
PATENT
WO 2014075168
PAPER

This work describes a scaffold hopping exercise that begins with known imidazo[1,2-a]pyrazines, briefly explores pyrazolo[1,5-a][1,3,5]triazines, and ultimately yields pyrazolo[1,5-a]pyrimidines as a novel class of potent TTK inhibitors. An X-ray structure of a representative compound is consistent with 11/2 type inhibition and provides structural insight to aid subsequent optimization of in vitro activity and physicochemical and pharmacokinetic properties. Incorporation of polar moieties in the hydrophobic and solvent accessible regions modulates physicochemical properties while maintaining potency. Compounds with enhanced oral exposure were identified for xenograft studies. The work culminates in the identification of a potent (TTK Ki = 0.1 nM), highly selective, orally bioavailable anticancer agent (CFI-402257) for IND enabling studies.
Discovery of Pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a Potent, Selective, Bioavailable Anticancer Agent
† Campbell Family Institute for Breast Cancer Research, University Health Network, TMDT East Tower, MaRS Centre, 101 College Street, Toronto, Ontario M5G 1L7, Canada
‡ Campbell Family Cancer Research Institute, University Health Network, Princess Margaret Cancer Center, 610 University Avenue, Toronto, Ontario M5G 2C4, Canada
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.5b00485
*E-mail: henry.pauls@cogeco.ca. Phone: 905-337-3446.
REFERENCES
Discovery of Pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a Potent, Selective, Bioavailable Anticancer AgentYong Liu, Radoslaw Laufer, Narendra Kumar Patel, Grace Ng, Peter B. Sampson, Sze-Wan Li, Yunhui Lang, Miklos Feher, Richard Brokx, Irina Beletskaya, Richard Hodgson, Olga Plotnikova, Donald E. Awrey, Wei Qiu, Nickolay Y. Chirgadze, Jacqueline M. Mason, Xin Wei, Dan Chi-Chia Lin, Yi Che, Reza Kiarash, Graham C. Fletcher, Tak W. Mak, Mark R. Bray, and Henry W. Pauls
Publication Date (Web): May 6, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.5b00485
////TTK inhibitors, CFI-402257, pyrazolo[1,5-a]pyrimidines, 11/2 type inhibitors, 1610759-22-2, 1610677-37-6
C[C@]1(O)C[C@H](C1)CNc2cc(nc3c(cnn23)c5ccc(C(=O)NC4CC4)c(C)c5)Oc6cccnc6
No comments:
Post a Comment