OR
ULIXERTINIB
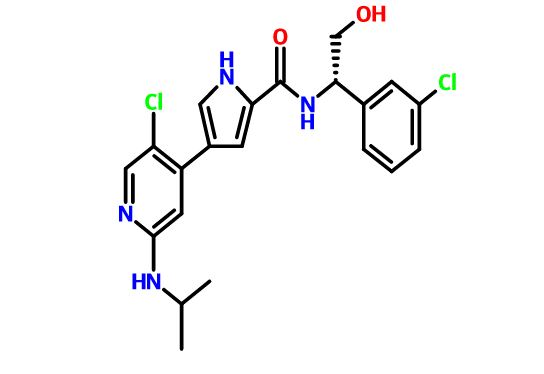
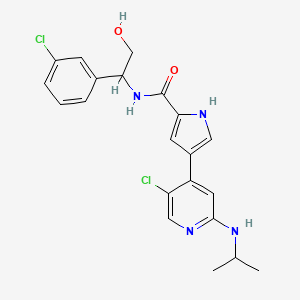
4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide
| Molecular Formula: | C21H22Cl2N4O2 |
|---|---|
| Molecular Weight: | 433.33098 g/mol |
BVD-523; BVD-ERK; BVD-ERK/HM; BVD-ERK/ST; VRT-0752271; VRT-752271; VX-271, V
уликсертиниб , أوليكسيرتينيب , 优立替尼 ,
4-[5-chloro-2-(isopropylamino)-4-pyridyl]-N-[(1S)-1-(3-chlorophenyl)-2-hydroxy-ethyl]-1H-pyrrole-2-carboxamide
CAS 869886-67-9
Vertex Pharmaceuticals, Incorporated innovators
ULIXERTINIB HCl

| Molecular Weight | 469.79 |
| Formula | C21H22Cl2N4O2●HCl |
| CAS | 1956366-10-1 |
| Chemical Name | 1H-Pyrrole-2-carboxamide, 4-[5-chloro-2-[(1-methylethyl)amino]-4-pyridinyl]-N-[(1S)-1-(3-chlorophenyl)-2-hydroxyethyl]-,hydrochloride(1:1) |
Ulixertinib malonate
4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib malonate)
- Originator Vertex Pharmaceuticals
- Developer BioMed Valley Discoveries
- Class Aminopyridines; Antineoplastics; Pyrroles; Small molecules
- Mechanism of Action Mitogen activated protein kinase 3 inhibitors; Mitogen-activated protein kinase 1 inhibitor
Highest Development Phases
- Phase I/II Acute myeloid leukaemia; Cancer; Myelodysplastic syndromes
- Phase I Pancreatic cancer
Most Recent Events
- 01 Mar 2016 Phase-I clinical trials in Pancreatic cancer (Combination therapy, First-line therapy, Metastatic disease) in USA (PO) (NCT02608229)
- 23 Nov 2015 BioMed Valley Discoveries and Washington University School of Medicine plan a phase Ib trial for Pancreatic cancer (First-line therapy, Metastatic disease, Combination therapy) (PO) (NCT02608229)
- 01 Nov 2014 Phase-I/II clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) and Myelodysplastic syndromes (Second-line therapy or greater) in USA (NCT02296242) (PO)

INTRODUCTION
Ulixertinib is in phase I/II clinical trials for the treatment of acute myelogenous leukemia (AML), myelodysplasia and advanced solid tumors.
Members of the family of B-cell CLL/lymphoma 2 proteins (BCL-2) are apoptosis regulators. These proteins control mitochondrial outer
membrane permeabilization (MOMP). Expression of BCL-2 protein blocks cell death in response to various cellular injuries. A number of cancers, including melanoma, breast, prostate, chronic lymphocytic leukemia, and lung cancer, may be caused by damage to the BCL-2 gene. Mutations in BCL-2 may also be a cause of resistance to cancer treatments. Unfortunately, resistance can quickly develop using conventional BCL-2 inhibitor therapies to treat cancer.
Extracellular-signal-regulated kinases (ERKs) are protein kinases that are involved in cell cycle regulation, including the regulation of meiosis, mitosis, and postmitotic functions in differentiated cells. Disruption of the ERK pathway is common in cancers. However, to date, little progress has been made developing effective ERK inhibitors for the treatment of cancer.
As the understanding of the molecular basis of cancer grows, there is an increased emphasis on developing drugs that specifically target particular nodes in pathways that lead to cancer. In view of the deficiencies noted above, there is, inter alia, a need for effective molecularly targeted cancer treatments, including combination therapies. The present invention is directed to meeting these and other needs.

Mitogen-activated protein kinase (MAPK) pathways mediate signals which control diverse cellular processes including growth, differentiation, migration, proliferation and apoptosis. One MAPK pathway, the extracellular signal-regulated kinase (ERK) signaling pathway, is often found to be up-regulated in tumors. Pathway members, therefore, represent attractive blockade targets in the development of cancer therapies (Kohno and Pouyssegur, 2006). For example, U.S. Patent No. 7,354,939 B2 discloses, inter alia, compounds effective as inhibitors of ERK protein kinase. One of these compounds, 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide, is a compound according to formula (I):

Pharmaceutical compositions are often formulated with a crystalline solid of the active pharmaceutical ingredient (API). The specific crystalline form of the API can have significant effects on properties such as stability and solubility / bioavailability. Instability and solubility characteristics can limit the ability to formulate a composition with an adequate shelf life or to effectively deliver a desired amount of a drug over a given time frame (Peterson et al., 2006).
Synergistic combination comprising an ERK1/2 inhibitor (such as ulixertinib) and a BCL-2 family inhibitor (such as navitoclax), assigned to BioMed Valley Discoveries (BVD), naming Decrescenzo and Welsch. BVD, presumably under license from Vertex, is developing ulixertinib (phase 2 trial), a small-molecule ERK 1/2 inhibitor for treating cancers including acute myelogenous leukemia and myelodysplastic syndrome. In June 2015, clinical data were presented at the 51st ASCO meeting in Chicago, IL.
BIOMED VALLEY DISCOVERIES

PATENT
WO2005113541 PDT PATENT
I-9 COMPD
SEE BELOW
PATENT
Novel crystalline forms of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib) can be prepared which exhibit improved properties, eg surprisingly improved stability and solubility characteristics. Also claimed is their use for treating cancer.
EXAMPLE 2
Preparation of Crystaline Free Base 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base was prepared according to the following synthesis scheme.
Stepl

C5H2CIFIN
257.43 C8H10CIIN2
ASYM-11 1606 296.54
ASYM-1 12060

ASYM-111938 ASYM-112393

ASYM-1 11935
In Step 1 , a clean and dry 200 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. Anhydrous ethanol (49.90 kg) was charged into the 200 L glass-lined reactor. ASYM-1 1 1606 (Asymchem) (12.70 kg) and isopropylamine (29.00 kg) were added into the mixture in turn. The mixture was heated to 65-75°C for refluxing. The mixture reacted at 65-75°C. After 20 h, the reaction was sampled and analyzed by HPLC every 4-6 h until the content of ASYM-1 1 1606 was <1 %. The mixture was cooled to 40-45°C and was concentrated at <45°C under reduced pressure (<-0.08 MPa) until 13-26 Lremained. The organic phase was washed with a sodium chloride solution and was stirred for 20-30 min and settled for 20-30 min before separation. The organic phase was concentrated at <30°C under reduced pressure (<-0.06 MPa) until 13-20 L remained. Petroleum ether (8.55 kg) was added into the concentrated mixture. The mixture was transferred into a 20 L rotary evaporator and continued concentrating at <30°C under reduced pressure (<-0.06 MPa) until 13-20 L remained. Then petroleum ether (8.55 kg) was added into the concentrated mixture. The mixture was cooled to 0-5°C and stirred for crystallization. After 1 h, the mixture was sampled and analyzed by wt% every 1 -2 h until the wt% of the mother liquor was <1 1 % or the change of the wt% between consecutive samples was <1 %. The mixture was filtered with a 10 L filter flask. The filter cake was sampled and analyzed for purity by HPLC. 10.50 kg of product was recovered as a brownish yellow solid at 99.39% purity.
In Step 2, a clean and dry 300 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. Glycol dimethyl ether (73.10 kg) was charged into the 300 L glass-lined reactor at 20-30°C. ASYM-1 12060 (Asymchem) (10.46 kg) and ASYM-1 1 1938 (Asymchem) (12.34 kg, 1 1 .64 kg after corrected) were added into the mixture in turn under the protection of nitrogen. Maintaining the temperature at 20-30°C, purified water (10.50 kg) and anhydrous sodium carbonate (5.67 kg) were added into the mixture. Palladium acetate (0.239 kg) and tricyclohexylphosphonium tetrafluoroborate (0.522 kg) were added into the mixture under the protection of nitrogen. After addition, the mixture was evacuated to <-0.06 MPa, and then filled with nitrogen to normal pressure. This was repeated for ten times until residual oxygen was <300 ppm. The mixture was heated to 75-85°C for refluxing. The mixture reacted at 75-85°C. After 4 h, the mixture was sampled and analyzed by HPLC every 2-3 h for content of ASYM-
1 12060. The content of AS YM-1 12060 was 6.18%, so additional ASYM-1 1 1938 (0.72 kg) was added and continued reaction until the content of ASYM-1 12060 was <3%. The mixture was cooled to 25-35°C and filtered with a 30 L stainless steel vacuum filter. The filter cake was soaked and washed twice with THF (14.10kg). The filtrate and washing liquor were combined and concentrated at <50°C under reduced pressure (<-0.08 MPa) until 10-15 L remained. The mixture was cooled to 15-25°C. Methanol (1 1 .05 kg) was added into the concentrated mixture. Then the mixture was stirred for crystallization. After 2 h, the mixture was sampled and analyzed by HPLC every 2-4 h until the wt% of the mother liquor was <2%. The mixture was filtered with a 30 L stainless steel vacuum filter. The filter cake was soaked and washed twice with methanol (8.30 kg). The filter cake was transferred into a 50 L plastic drum. Then ethyl acetate (7.10 kg) and petroleum ether (46.30 kg) were added into the drum. The mixture was stirred for 1.5-2 h and then filtered with a nutsche filter. The filter cake was soaked and washed with petroleum ether (20.50 kg). The filter cake was dried in the nutsche filter under nitrogen at 30-40°C. After 8 h, the solid was sampled and Karl Fischer (KF) analysis was performed in intervals of 4-8 h to monitor the drying process. Drying was completed when the KF result was <1 .0% water. During drying, the solid was turned over and mixed every 4-6 h. 12.15 kg of product was recovered as a brownish yellow solid at 98.32% purity.
In Step 3, a clean and dry 300 L glass-lined reactor was evacuated to <-0.08 MPa, and then filled with nitrogen to normal pressure three times. THF (62.58 kg) was charged into the 300 L glass-lined reactor at 15-30°C. Then the stirrer was started. ASYM-1 12393 (12.00 kg, 1 1 .70 kg after corrected) was added into the mixture. The mixture was stirred until the solid dissolved completely. Maintaining the temperature at 15-30°C, a lithium hydroxide solution which was
prepared with lithium hydroxide monohydrate (5.50 kg) in purified water (70.28 kg) was added into the mixture. Then diethylamine (3.86 kg) was added. The mixture was heated to 60-70°C for refluxing. The mixture reacted at 60-70°C. After 30 h, the reaction was sampled and analyzed by HPLC every 4-6 h until the content of intermediate at relative retention time (RRT)=1 .39-1 .44 was <1 % and the content of ASYM-1 12393 was <1 %. HPLC conditions for this analysis are set forth in Table 1 .
Table 1 : HPLC Parameters

The mixture was cooled to 25-35°C and MTBE (25.97 kg) was added into the mixture. The mixture was stirred for 20-30 min and filtered via an in-line fluid filter. The filtrate was transferred into a 300 L glass-lined reactor and settled for 20-30 min before separation. The pH of the obtained aqueous phase was adjusted with a 6 N hydrochloric acid solution which was prepared from concentrated hydrochloric acid (14.86 kg) in purified water (10.88 kg) at the rate of 5-8 kg/h at 15-25°C until the pH was 1 -2. The pH of the mixture was adjusted again with a saturated sodium carbonate solution which was prepared from sodium carbonate (5.03 kg) in purified water (23.56 kg) at the rate of 3-5 kg/h at 15-25°C until the pH was 6.4-6.7. Then the pH of the mixture was adjusted with a hydrochloric acid solution which was prepared from concentrated hydrochloric acid (1 .09 kg) in purified water (0.80 kg) until the pH was 6.2-6.4. The mixture was filtered with a nutsche filter. The filter cake was transferred into a 300 L glass-lined reactor and purified water (1 17.00 kg) was added. The mixture was stirred and sampled and analyzed by HPLC until the p-toluenesulfonic acid residue of the filter cake was <0.5%. Then the mixture was filtered. The filter cake was dried in the tray drier under nitrogen at 55-65°C until KF<10%. The solid and MTBE (8.81 kg) were charged into a 50 L stainless steel drum. The mixture was stirred for 1 -2 h. The mixture was filtered with a 30 L stainless steel vacuum filter. The filter cake was dried in the nutsche filter at 50-60°C. After 8 h, the solid was sampled and analyzed by KF every 4-8 h until KF<5%. During drying, the solid was turned over and mixed every 4-6 h. 6.3 kg of product was recovered as an off-white solid at 98.07% purity.
In Step 4, a dry and clean 50 L flask was purged with nitrogen for 20 min. DMF (30.20 kg) was charged into the 50 L flask reactor. Then the stirrer was started. Maintaining the temperature at 15-25°C, ASYM-1 12394 (3.22 kg, 2.76 kg after corrected) was added into the mixture. The mixture was stirred until the solid dissolved completely. The mixture was cooled to -10 to -20°C and 1 -hydroxybenzotriazole hydrate (2.10 kg) was added into the mixture at -10 to -20°C. Then EDCI (2.41 kg) was added into the mixture in five portions at an interval of about 5-10 min. The mixture was cooled to -20 to -30°C and ASYM-1 1 1888 (Asymchem) (1 .96 kg) was added into the mixture at -20 to -30°C. Then DIEA (1 .77 kg) was added into the mixture at the rate of 3-4 kg/h. The mixture was heated to 15-25°C at the rate of 5-10°C/h. The mixture was reacted at 15-25°C. After 6-8 h, the mixture was sampled and analyzed by HPLC every 2-4 h until the content of ASYM-1 12394 was <2%. The mixture was cooled to 0-10°C and the reaction mixture was quenched with a solution which was prepared from ethyl acetate (28.80 kg) in purified water (12.80 kg) at 0-10°C. The mixture was extracted three times with ethyl acetate (28.80 kg). For each extraction the mixture was stirred for 20-30 min and settled for 20-30 min before separation. The organic phases were combined and washed twice with purified water (12.80 kg). The mixture was stirred for 20-30 min and settled for 20-30 min before separation for each time. Then the obtained organic phase was filtered through an in-line fluid filter. The filtrate was transferred into a 300 L glass-lined reactor. The mixture was washed twice with a 5% acetic acid solution, which was prepared from acetic acid (2.24 kg) in purified water (42.50 kg). The solution was added at the rate of 10-20 kg/h. The organic phase was washed twice with a sodium carbonate solution, which was prepared from sodium carbonate (9.41 kg) in purified water (48.00 kg). The organic phase was washed twice with a sodium chloride solution, which was prepared from sodium chloride (16.00 kg) in purified water (44.80 kg). The organic phase was transferred into a 300 L glass-lined reactor. Anhydrous sodium sulfate (9.70 kg) was added into the mixture and the mixture was stirred for 2-4 h at 15-30°C. The mixture was filtered with a nutsche filter, which was pre-loaded with about 1 cm thick silica gel (7.50 kg). The filter cake was soaked and washed with ethyl acetate (14.40 kg) before filtration. The filtrates were combined and the combined filtrate was added into a 72 L flask through an in-line fluid filter. The mixture was concentrated at T≤40°C under reduced pressure (P<-0.08 MPa) until 3-4 L remained. Then MTBE (4.78 kg) was added into the mixture. The mixture was cooled to 0-10°C for crystallization with stirring. After 1 h, the mixture was sampled and analyzed by wt% every 1-2 h until the wt% of the mother liquor was <5% or the change of wt% between consecutive samples was <1%. The mixture was filtered with a vacuum filter flask and the filter cake was dried in the tray drier under nitrogen at 30-40°C until KF<0.5%. 3.55 kg of product was recovered as an off-white solid at 100% purity.
EXAMPLE 3A
Preparation of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C

4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C was prepared from 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base as follows. ASYM-1 1 1935 (10.4 kg) was added to a stirred mixture of anhydrous ethanol (73.9 kg), methanol (4.1 kg) and isopropanol (4.1 kg). The mixture was heated to 70-75°C and stirred until all the solids dissolved. Anhydrous HCI (37 wt%, 1 .1 eq) in a mixture of ethanol/methanol/isopropanol (90:5:5) was added and the mixture maintained at 70-75°C for 2 hours after the addition was completed. The mixture was then cooled to 15-25°C at a rate of 5-15°C per hour and stirred at this temperature until the desired polymorphic purity was reached. The end point of the crystallization/polymorph conversion was
determined by the absence of an XRPD peak at about 10.5° 2Θ in three successive samples.
The mixture was then filtered and washed successively with a pre-prepared solution of anhydrous ethanol (14.8 kg), methanol (0.8 kg) and isopropanol (0.8 kg), followed by MTBE (2 x 21 kg). Avoidance of delay in the washing of the filter cake is preferable because the polymorph may be unstable in the wet filter cake in the presence of reagent alcohol and improved stability was observed after the MTBE wash has been performed. The wet filter cake was then dried in a heated filter funnel or a tray drier at 40-50°C until dry. Typical yields were about 85-90%.
EXAMPLE 3B
Alternative Preparation of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C

ASYM-1 15985
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C was also prepared from 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide free base as follows. A dry and clean 72 L flask was purged with nitrogen for 20 min. Anhydrous ethanol (21 .35 kg) methanol (1 .17 kg) and isopropanol (1 .19 kg) were charged into the 72 L flask at 15-25°C and the mixture was stirred for 20-30 min. ASYM-1 1 1935 (3.01 kg) was added into the mixture and heated to 70-75°C at the rate of 15-25°C/h and stirred until the solid dissolved completely.
An alcohol / HCI solution was prepared as follows. Anhydrous ethanol (1.500 kg) methanol (0.088 kg) and isopropanol (0.087 kg) were charged into a 5 L flask at 15-25°C and the mixture was stirred for 20-30 min. The mixture was bubbled with hydrogen chloride through a dip tube under stirring at 10-25°C. After 2 h, the mixture was sampled and analyzed every 2-4 h until the wt% of hydrogen chloride was > 35%.
The alcohol / HCI solution (0.519 kg) prepared above was added dropwise into the mixture at the rate of 0.5-1.0 kg/h at 70-75°C. Seed crystal (0.009 kg) was added into the mixture and the alcohol / HCI solution (0.173 kg) prepared above was added into the mixture at the rate of 0.5-1 .0 kg/h at 70-75°C. After addition, the mixture was stirred for 1 -2 h at 70-75°C. The mixture was cooled to 15-25°C at the rate of 5-15°C/h and stirred for 4-6 h. The mixture was heated to 70-75°C at the rate of 15-25°C/h and stirred for 8-10 h at 70-75°C. The mixture was cooled to 15-25°C at the rate of 5-15°C/h and stirred for 4-6 h. The mixture was filtered with a vacuum filter flask. The filter cake was soaked and rinsed with a solution which was prepared from anhydrous ethanol (4.25 kg) and methanol (0.24 kg) and isopropanol (0.24 kg) before filtration. The filter cake was dried in a drying room under nitrogen at 40-50°C until the ethanol residue was <0.5% and methanol residue was <0.3% and isopropanol residue was <0.3%. 2.89 kg of product was recovered as a white solid at 99.97% purity.
PATENT
WO-2016123581
Novel crystalline malonate salt forms of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-1H-pyrrole-2-carboxylic acid[1-(3-chlorophenyl)-2-hydroxyethyl]amide (referred to as ulixertinib malonate) and composition comprising them. Also claimed is their use for treating cancer.
EXAMPLE 6
Aqueous Disolution of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1-(3-chlorophenyl)-2-hydroxyethyl]amide Malonate Form A
Samples of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C and 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2 -carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide malonate Form A were each shaken at ambient temperature in fasting state simulated gastric fluid (FaSSGF) pH 1.6 for 30 minutes. Concentration of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide was measured at 5, 15 and 30 minutes.
After 30 minutes, the samples were removed from FaSSGF, placed in fasting state simulated intestinal fluid (FaSSIF) pH 6.5, with shaking, for an additional 5 hours. Concentration of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide was measured at 10, 30, 60 90, 120, 180, 270, and 300 minutes. Results are summarized in Table 13 and shown in FIG. 10A (FaSSGF) and FIG. 10B (FaSSIF).
Table 13: Solubility of 4-(5-Chloro-2-isopropylaminopyridin-4-yl)-1 H-pyrrole-2-carboxylic acid [1 -(3-chlorophenyl)-2-hydroxyethyl]amide Form C and Malonate Form A.

PATENT
PATENT
WO2015095834
PATENT
Example 1 Compound 1-9 was prepared as follows:

1-9
2,2,2-TrichIoro-l-(4-iodo-lH-pyrrol-2-yl)ethanone: To a stirred solution of 50 g (235 mmol, 1.0 equiv.) of 2,2,2-trichloro-l-(lH-pyrrol-2-yl)-ethanone in dry dichloromethane (400 mL) under nitrogen, a solution of iodine monochloride (39 g, 240 mmol, 1.02 equivalents) in of dichloromethane (200 mL) was added dropwise. The resulting mixture was stirred at room temperature for 2 hours. The solution was washed with 10% potassium carbonate, water, 1.0 M sodium thiosulfate, saturated sodium chloride, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The solid was recrystallized from hexanes/methyl acetate to afford the title compound (68.5g, 86%) as a colorless solid (86%). MS FIA: 335.8, 337.8 ES-.
4-Iodo-lH-pyrrole-2-carboxyIic acid methyl ester: To a stirred solution of 2,2,2- trichloro-l-(4-iodo-lH-pyrrol-2-yl)ethanone (68g, 201 mmol, 1.0 equivalent) in dry methanol (400 mL) under nitrogen, was added a solution of sodium methoxide in methanol (4.37 M, 54 mL, 235 mmol, 1.2 equivalents) over 10 minutes. The resulting mixture was stirred at room temperature for 1 hour. The volatiles were removed under reduced pressure and the crude was then partitioned between water and tert- butylmethyl ether. The organic phase was separated, washed two times with water, saturated sodium chloride, dried over sodium sulfate, filtered and concentrated under vacuum to afford the title compound (48g, 96%) as a colorless solid, that was used directly without further purification.
4-Iodo-l-(toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester: 4-Iodo- lH-pyrrole-2-carboxylic acid methyl ester (24.6 g, 98 mmol, 1.0 equivalent) was dissolved in dichloromethane (150 mL) and triethylamine (30 mL, 215.6 mmol, 2.2 equivalents). 4-(Dimethylamino)pyridine (1.2 g, 9.8 mmol, 0.1 equivalent) and p- toluenesulfonylchloride (20.6 g, 107.8 mmol, 1.1 equivalents) were added and the reaction mixture was stirred for 16 hours at room temperature. The reaction was quenched with 1 M ΗC1 and the organic layer was washed with aqueous sodium bicarbonate and brine. After drying over magnesium sulfate, the solvent was removed under reduced pressure and the residue was crystallized from tert-butylmethyl ether, yielding the title compound as a pale yellow solid (30 g, 75%). Rt(min) 8.259 minutes.
4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yI)-l-(toluene-4-sulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester: To a degassed solution of 4-iodo-l- (toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (20 g, 49.4 mmol, 1.0 equivalent) and bis(pinacolato)diborane (15 g, 65 mmol, 1.3 equivalents) in DMF (200 mL) under nitrogen, was added dichloro[l,l '- bis(diphenylphosphino)ferrocene]palladium (II) dichloromethane adduct (3.6 g, 4.9 mmol, 0.1 equivalent). The reaction mixture was then stirred at 80 °C for 18 hours. After removing the DMF under reduced pressure, the resulting thick oil residue was suspended in diethyl ether (500 mL) and a solid precipitated immediately. This solid was removed by filtration and the filtrate was washed with IM HCl, water, brine and dried over MgS0 . Concentration afforded the title compound as a white solid and used without further purification (10 g, 50%). LC/MS: Rt(min) 4.6; 406.4 ES+. MS FIA: 406.2 ES+. 'pfNMR δ 1.2 (s, 12H), 2.35 (s, 3H), 3.8 (s, 3H), 7.2 (m, 3H), 7.8 (d, 2H), 8.0 (s, IH).
N,N'-2-(5-Chloro-4-iodo-pyridyI)-isopropyIarnine:
Method A. (Microwave)
In a 10 mL microwave tube, 5-chloro-2-fluoro-4-iodopyridine (1.0 g, 3.9 mmol, 1.0 equivalent) was dissolved in DMSO (4.0 mL) and then ispropylamine (0.99 mL, 11.7 mmol, 3.0 equivalents) was added. The tube was sealed and placed under microwave irradiation for 600 sec at 150 °C. This reaction was repeated six times. The reaction mixtures were combined, then diluted in ethyl acetate and washed with water. After drying over sodium sulfate, the solvent was evaporated to afford the title compound as a thick brown oil (5.6 g, 80% ) which was used directly without further purification. Rt(min) 4.614; MS FIA: 296.9 ES+. 'pfNMRsssssss δ 1.25 (d, 6H), 3.65 (m, IH), 7.15 (s, IH), 7.75 (s, IH).
Method B: (Thennal)
5-Chloro-2-fluoro-4-iodopyridine (400 mg, 1.55 mmol, 1.0 equivalent) was dissolved in ethanol (5.0 mL) and then isopropylamine (0.66 mL, 7.8 mmol, 5.0 equivalents) was added. The resulting solution was stirred at 80 °C for 48 hours. The reaction mixture was then diluted in ethyl acetate and washed with water. After drying over sodium sulfate, the solvent was evaporated and a thick brown oil was obtained, which was then purified by flash chromatography on silica gel eluting with mixtures of hexanes/ethyl acetate (from 99:1 to 80:20) to afford the title compound as a pale yellow solid (96 mg, 21%).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-l-(toluene-4-suIfonyl)-lH-pyrrole-2- carboxylic acid methyl ester: To a solution of N,N'-2-(5-chloro-4-iodo-pyridyl)- isopropylamine (0.53 g, 1.8 mmol, 1.0 equivalent) and 4-(4,4,5,5-tetramethyl- [l,3,2]dioxaborolan-2-yl)-l-(toluene-4-sulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (0.78 g, 1.8 mmol, 1.0 equivalent) in DME (4.0 mL) was added a solution of aqueous 2 M sodium carbonate (1.0 mL) followed by Pd(PPh3)4 (0.21 mg, 0.18 mmol, 0.1 equivalent). The microwave tube was sealed and the reaction mixture was irradiated by microwave for 1800 sec. at 170 °C. The cmde of six reactions were combined and diluted in ethyl acetate and washed with water. After drying the organic layer with sodium sulfate, the solvent was removed and the resulting thick oil was adsorbed on silica gel. The crude was then purified by flash chromatography on silica, eluting with hexanes/ethyl acetate mixtures (from 99:1 to 70:30) to afford the title compound as a yellow solid (3.1 g, 61% over two steps). Rt(min) 6.556. MS FIA: 448.1 ES+. 'HNMR δ 1.45 (d, 6H), 2.5 (s, 3H), 3.81 (s, 3H), 6.8 (s, IH), 7.35 (s, IH),
7.4 (d, 2H), 8.0 (m ,3H), 8.3 (s, IH).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-l-(2,4,6-trimethylbenzenesulfonyl)- lH-pyrrole-2-carboxylic acid methyl ester: To a solution of N,N'-2-(5-chloro-4- iodo-pyridyl)-isopropylamine (96 mg, 0.32 mmol, 1.0 equivalent) and 4-(4,4,5,5- tetramethyl-[ 1 ,3,2]dioxaborolan-2-yl)- 1 -(2,4,6-trimethylbenzenesulfonyl)- lH-pyrrole- 2-carboxylic acid methyl ester (152 mg, 0.35 mmol, 1.1 equivalents) in DME (2 mL), was added a solution of aqueous 2 M sodium carbonate (0.2 mL) followed by Pd(PPh ) (37 mg, 0.032 mmol, 0.1 equivalent). The reaction mixture was stirred at 80 °C for 16 hours. The crude was diluted in ethyl acetate and washed with water. After drying the organic layer with sodium sulfate, the solvent was removed and the resulting thick oil was adsorbed on silica gel. The cmde was then purified by flash chromatography on silica, eluting with hexanes/ethyl acetate mixtures (from 99:1 to 80:20) to afford the title compound as a yellow solid (65 mg, 43%). Rt(min) 7.290. MS FIA:476.1 ES+.
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxyIic acid:
Method A. (Microwave)
A solution of 4-(5-chloro-2-isopropylaminopyridin-4-yl)-l-(toluene-4-sulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester (3.1 g, 6.9 mmol, 1.0 equivalent) in TΗF (2.0 mL) was added to a solution of lithium hydroxide monohydrated (710 mg, 17.3 mmol,
2.5 equivalents) in water (3.0 mL). The microwave tube was sealed and the reaction mixture was irradiated by microwave for 1200 sec. at 150 °C. The cmde solution was acidified with aqueous 6Ν ΗC1. The solvent was evaporated off to afford the title compound which was used directly without further purification. Rt(min): 3.574. FIA MS: 279.9 ES+; 278.2 ES-.
Method B: (Thermal)
A solution of 4-(5-chloro-2-isopropylaminoρyridin-4-yl)-l-(2,4,6- trimethylbenzenesulfonyl)-lH-pyrrole-2-carboxylic acid methyl ester (0.69 g, 1.4 mmol, 1.0 equivalent) in TΗF (3.0 mL) was added to a solution of lithium hydroxide monohydrated (1.19 g, 29 mmol, 20.0 equivalents) in water (3.0 mL). The mixture was then refluxed for 8 hours. The cmde solution was acidified with aqueous 6N ΗC1 until cloudy, the organic solvent was partially removed and the product precipitated. The title compound was isolated by filtration and washed with water and diethyl ether, yielding a white solid (0.38 g, 96%).
4-(5-Chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxyIic acid [l-(3- ch!orophenyl)-2-hydroxyethyl] amide: To a suspension of 4-(5-chloro-2- isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxylic acid (1.93 g, 6.9 mmol, 1.0 equivalent) in DMF (5.0 mL) was added EDCI (1.45 g, 7.6 mmol, 1.1 equivalents), ΗOBt (0.94 g, 6.9 mmol, 1.0 equivalent) and (5)-3-chlorophenylglycynol (1.58 g, 7.6 mmol, 1.1 equivalents). Dusopropylethylamme (2.7 mL) was then added and the resulting mixture was stirred a room temperature overnight. The mixture was then poured into water and extracted with ethyl acetate. After drying over sodium sulfate, the solvent was removed and the crude was adsorbed on silica gel. Purification was effected by flash chromatography on silica, eluting with mixtures of hexanes/acetone (from 80:20 to 60:40) to afford the title compound as white solid (1.9 g, 64%). Rt(min) 4.981s. FIA MS: 433.1 ES+; 431.2 ES-. 1ΗNMR (CD3OD) δ 1.31 (d, 6H), 3.85 (m, 3H), 5.15 (t, IH), 7.01 (s, IH), 7.25 (m, 3H), 7.4 (s, IH), 7.45 (s, IH), 7.7 (s, IH), 7.95 (s, IH).
Example 2 Compound 1-9 was also prepared according to following alternate method:

2,5-DichIoro-4-nitropyridine N-oxide: To a suspension of 2-chloro-5-chloropyridine (10 g, 0.067 mol) in acetic anhydride (25 mL) was added hydrogen peroxide 30% (25 mL) in small portions. This mixture was stirred at room temperature for 24 hours and then heated at 60 °C for 30 hours. After removing the excess of acetic acid under reduced pressure, the residue was added in small portions to concentrated sulfuric acid (15 mL). The resulting solution was added to a mixture of concentrated sulfuric acid (15 mL) and fuming nitric acid (25 mL) and then heated at 100 °C for 90 minutes. The reaction mixture was poured on ice, neutralized with solid ammonium carbonate and finally with aqueous ammonia until a basic pH was obtained and. A precipitate formed. The precipitate was collected by filtration to afford the title compound as a pale yellow solid (3.1 g), Rt(min) 3.75. MS FIA shows no peak. 'pfΝMR (DMSO-de) δ 8.78 (s, IH), 9.15 (s, IH).
4-Bromo-2-chloro-5-N-isopropylpyridin-2-amine N-oxide: To 2,5-dichloro-4- nitropyridine Ν-oxide (400 mg, 1.9 mmol) was added acetyl bromide (2 mL) very slowly. The reaction mixture was then heated at 80 °C for 10 minutes. The solvent was removed under a stream of nitrogen and the cmde product was dried under high vacuum. The cmde material (165 mg, 0.62 mmol) was dissolved in ethanol (2 mL), z'so-propylamine (0.53 mL) added and the resulting mixture was heated at 80 °C for 2 hours. The cmde solution was then purified by reversed phase HPLC (acetonitrile/water/TFA 1%) to afford the title compound as a pale yellow solid (60 mg, 36.6%). Rt(min) 5.275. MS FIA264.8, 266.9 ES+.
4-(5-chloro-2-isopropylaminopyridin-4-yl)-lH-pyrrole-2-carboxylic acid [l-(3- chlorophenyl)-2-hydroxyethyl] amide (1-9): 4-Bromo-2-chloro-5-N- isopropylpyridin-2-amine N-oxide (25 mg, 0.094 mmol, 1.0 equivalent) and 4- (4,4,5, 5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-l-(2,4,6-trimethylbenzensulfonyl)-lH- pyrrole-2-carboxylic acid methyl ester (39 mg, 0.094 mmol, 1.0 equivalent) were dissolved in benzene (5 mL) then aqueous 2M Νa2C03 (1 mL) and Pd(PPh3)4 (115.6 mg, 0.1 mmol, 0.2 equivalent) were added and the resulting suspension was heated at reflux at 80 °C for 16 hours. The reaction mixture was diluted in ethyl acetate, washed with water and dried over anhydrous sodium sulfate to afford 4-(5-chloro-2- isopropylamino-pyridin-4-yl)- 1 -(2,4,6-trimethyl-benzenesulfonyl)- lH-pyrrole-2- carboxylic acid methyl ester N-oxide (R (min) 6.859. MS FIA: 492.0 ES+) which was then treated with a 2 M solution of PC13 in dichloromethane (1 mL) at room temperature. After 10 minutes, the solvent was removed under a stream of nitrogen and the cmde oil was dissolved in methanol (1 mL) and aqueous 1 M ΝaOΗ (1 mL). The resulting mixture was heated at reflux for 16 hours then the cmde solution was acidified using aqueous 1 M ΗC1 and the solvent was removed. The resulting 4-(5- chloro-2-isopropylamino-pyridin-4-yl)-lΗ-pyrrole-2-carboxylic acid (R (min) 3.527. MS FIA: 279.4 ES+; 278.2 Es-) was suspended in DMF (3 mL) together with EDCI (36 mg, 0.19 mmol, 2 equivalents), HOBt (26 mg, 0.19 mmol, 2 equivalents), (S)-3- chlorophenylglycinol HCl salt (59 mg, 0.28 mmol, 3 equivalents) and DIEA (0.12 mL, 0.75 mmol, 8 equivalents). The resulting mixture was stirred at room temperature for 16 hours. The reaction mixture was diluted in ethyl acetate, washed with water and dried over sodium sulfate. After removing the solvent under reduced pressure, the cmde product was purified by reversed phase HPLC (acetonitrile/water/TFA 1%) to afford the title compound as a white solid (4.8 mg, 8.1%).
PATENT
US20150512092015-02-19COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF MEK
US73549392008-04-08Pyrrole inhibitors of ERK protein kinase, synthesis thereof and intermediates thereto
Research scientist Tony Huang works in a laboratory at Vertex Pharmaceuticals Inc. in San Diego
REFERENCES
1 . Kohno M, Pouyssegur J (2006) Targeting the ERK signaling pathway in cancer therapy. Ann Med 38: 200-21 1 .
2. Kuby, J., Immunology, 3rd Ed., W.H. Freeman & Co., New York.
3. Lee DC, Webb ML(2003) Pharmaceutical Analysis. John Wiley & Sons, Inc., New York: 255-257.
4. Peterson ML, Hickey MB, Zaworotko MJ and Almarsson O (2006) Expanding the Scope of Crystal Form Evaluation in Pharmaceutical Science. J Pharm Pharmaceut Sci 9(3):317-326.
5. Pierce Catalog and Handbook, 1994-1995; Pierce Chemical Co., Rockford, III.
6. Remington, The Science and Practice of Pharmacy (21 st Edition, Lippincott Williams and Wilkins, Philadelphia, PA.
7. The United States Pharmacopeia-National Formulary, The United States Pharmacopeial Convention, Rockville, MD.

Gabriel Martinez-Botella
Director, Chemistry at Sage Therapeutics
Experience

Director, Chemistry
Sage Therapeutics

Principal Scientist, Team Leader
AstraZeneca

Sr Scientist
Vertex Pharmaceuticals
Education


///////////ULIXERTINIB, BVD-523; BVD-ERK, BVD-ERK/HM, BVD-ERK/ST, VRT-0752271, VRT-752271, VX-271, уликсертиниб ,أوليكسيرتينيب ,优立替尼 , PHASE 2, Vertex Pharmaceuticals, BioMed Valley Discoveries, UNII:16ZDH50O1U, 869886-67-9
CC(C)NC1=NC=C(C(=C1)C2=CNC(=C2)C(=O)NC(CO)C3=CC(=CC=C3)Cl)Cl

BEST PLACE TO BUY OXYCODONE ONLINE WITH AND WITHOUT PRESCRIPTION
ReplyDeleteHow should I take Adderall?Buy Adderall online here,Adderall discreetly shipped to your home,Adderall 30 mg,
BUY GREENSTONE-Xanax Online
Buy Xanax online
Buy Adderall Online, discreetly shipped to your home , Adderall on sale, where can I buy Adderall
BUY DEMEROL ONLINE WITHOUT PRESCRIPTION
DR EMU WHO HELP PEOPLE IN ANY TYPE OF LOTTERY NUMBERS
ReplyDeleteIt is a very hard situation when playing the lottery and never won, or keep winning low fund not up to 100 bucks, i have been a victim of such a tough life, the biggest fund i have ever won was 100 bucks, and i have been playing lottery for almost 12 years now, things suddenly change the moment i came across a secret online, a testimony of a spell caster called dr emu, who help people in any type of lottery numbers, i was not easily convinced, but i decided to give try, now i am a proud lottery winner with the help of dr emu, i won $1,000.0000.00 and i am making this known to every one out there who have been trying all day to win the lottery, believe me this is the only way to win the lottery.
Dr Emu can also help you fix this issues
(1)Ex back.
(2)Herbal cure & Spiritual healing.
(3)You want to be promoted in your office.
(4)Pregnancy spell.
(5)Win a court case.
Contact him on email Emutemple@gmail.com
What’s app +2347012841542
Website Https://emutemple.wordpress.com/
Facebook page Https://web.facebook.com/Emu-Temple-104891335203341