
Lenvatinib
For the treatment of patients with progressive radioiodine-refractory, differentiated thyroid cancer (RR-DTC).
CAS 417716-92-8,
CAS 857890-39-2 (lenvatinib mesylate)
E 7080, ER-203492-00, E7080, E 7080,
4-[3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6-quinolinecarboxamide
Molecular Formula: C21H19ClN4O4
Molecular Weight: 426.85296
Eisai Co., Ltd INNOVATOR
| Eisai R&D Management Co., Ltd. |

Lenvatinib was granted Orphan Drug Designation for thyroid cancer by the health authorities in Japan in 2012, and in Europe and the U.S in 2013. The first application for marketing authorization of lenvatinib in the world was submitted in Japan on June 2014. Eisai is planning to submit applications for marketing authorization in Europe and the U.S. in the second quarter of fiscal 2014. Lenvatinib is an oral multiple receptor tyrosine kinase (RTK) inhibitor with a novel binding mode that selectively inhibits the kinase activities of vascular endothelial growth factor receptors (VEGFR), in addition to other proangiogenic and oncogenic pathway-related RTKs including fibroblast growth factor receptors (FGFR), the platelet-derived growth factor (PDGF) receptor PDGFRalpha, KIT and RET that are involved in tumor proliferation. This potentially makes lenvatinib a first-in-class treatment, especially given that it simultaneously inhibits the kinase activities of FGFR as well as VEGFR.

LENVATINIB BASE
COSY PREDICT

| Systematic (IUPAC) name | |
|---|---|
| 4-[3-chloro-4-(cyclopropylcarbamoylamino)phenoxy]-7-methoxy-quinoline-6-carboxamide | |
| Clinical data | |
| Legal status | ℞ Prescription only |
| Identifiers | |
| CAS number | |
| ATC code | None |
| PubChem | CID 9823820 |
| ChemSpider | 7999567 |
| UNII | EE083865G2 |
| Chemical data | |
| Formula | C21H19ClN4O4 |
| Mol. mass | 426.853 g/mol |
Lenvatinib (E7080) is a multi-kinase inhibitor that is being investigated for the treatment of various types of cancer by Eisai Co. It inhibits both VEGFR2 and VEGFR3 kinases.[1] The substence was granted orphan drug status for the treatment of various types of thyroid cancer that do not respond toradioiodine; in the US and Japan in 2012 and in Europe in 2013[2] and is now approved for this use.

Clinical trials
Lenvatinib has had promising results from a phase I clinical trial in 2006[3] and is being tested in several phase II trials as of October 2011, for example against hepatocellular carcinoma.[4] After a phase II trial testing the treatment of thyroid cancer has been completed with modestly encouraging results,[5] the manufacturer launched a phase III trial in March 2011.[6]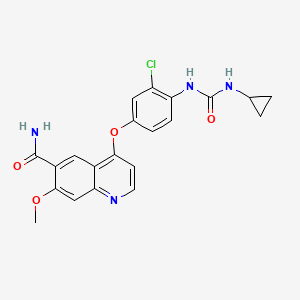
 Lenvatinib Mesilate
Molecular formula: C21H19ClN4O4,CH4O3S =523.0.
CAS: 857890-39-2.
UNII code: 3J78384F61.
Lenvatinib Mesilate
Molecular formula: C21H19ClN4O4,CH4O3S =523.0.
CAS: 857890-39-2.
UNII code: 3J78384F61.About the Lenvatinib (E7080) Phase II Study The open-label, global, single-arm Phase II study of multi-targeted kinase inhibitor lenvatinib (E7080) in advanced radioiodine (RAI)-refractory differentiated thyroid cancer involved 58 patients with advanced RAI refractory DTC (papillary, follicular or Hurthle Cell) whose disease had progressed during the prior 12 months. (Disease progression was measured using Response Evaluation Criteria in Solid Tumors (RECIST).) The starting dose of lenvatinib was 24 mg once daily in repeated 28 day cycles until disease progression or development of unmanageable toxicities.
2. About Thyroid Cancer Thyroid cancer refers to cancer that forms in the tissues of the thyroid gland, located at the base of the throat or near the trachea. It affects more women than men and usually occurs between the ages of 25 and 65. The most common types of thyroid cancer, papillary and follicular (including Hurthle Cell), are classified as differentiated thyroid cancer and account for 95 percent of all cases. While most of these are curable with surgery and radioactive iodine treatment, a small percentage of patients do not respond to therapy. 3. About Lenvatinib (E7080) Lenvatinib is multi-targeted kinase inhibitor with a unique receptor tyrosine kinase inhibitory profile that was discovered and developed by the Discovery Research team of Eisai's Oncology Unit using medicinal chemistry technology. As an anti-angiogenic agent, it inhibits tyrosine kinase of the VEGF (Vascular Endothelial Growth Factor) receptor, VEGFR2, and a number of other types of kinase involved in angiogenesis and tumor proliferation in balanced manner. It is a small molecular targeting drug that is currently being studied in a wide array of cancer types. 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (additional name: 4-[3-chloro-4-(N′-cyclopropylureido)phenoxy]-7-methoxyquinoline-6-carboxamide) is known to exhibit an excellent angiogenesis inhibition as a free-form product, as described in Example 368 of Patent Document 1. 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide is also known to exhibit a strong inhibitory action for c-Kit kinase (Non-Patent Document 1, Patent Document 2). However, there has been a long-felt need for the provision of a c-Kit kinase inhibitor or angiogenesis inhibitor that has high usability as a medicament and superior characteristics in terms of physical properties and pharmacokinetics in comparison with the free-form product of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide.
[Patent Document 1] WO 02/32872
[Patent Document 2] WO 2004/080462
[Non-Patent Document 1] 95th Annual Meeting Proceedings, AACR (American Association for Cancer Research), Volume 45, Page 1070-1071, 2004
............................. PATENT

http://www.google.co.in/patents/US8058474
EXAMPLES Examples will now be described to facilitate understanding of the invention, but the invention is not limited to these examples. Example 1Phenyl N-(2-chloro-4-hydroxyphenyl)carbamate



Example 44-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide In a reaction vessel were placed 7-methoxy-4-chloro-quinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethylsulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) in that order, under a nitrogen atmosphere. After stirring at 20° C. for 30 minutes, the temperature was raised to 65° C. over a period of 2.5 hours. After stirring at the same temperature for 19 hours, 33% (v/v) acetone water (5.0 L) and water (10.0 L) were added dropwise over a period of 3.5 hours. Upon completion of the dropwise addition, the mixture was stirred at 60° C. for 2 hours, and 33% (v/v) acetone water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or higher over a period of 1 hour. After then stirring at 40° C. for 16 hours, the precipitated crystals were collected by filtration using a nitrogen pressure filter, and the crystals were washed with 33% (v/v) acetone water (33.3 L), water (66.7 L) and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum drier to give 7.78 kg of the title compound (96.3% yield).
..............................
SYNTHESIS

1H NMR PREDICT


13 C NMR PREDICT


.......................
PATENThttp://www.google.co.in/patents/US7253286
EX 368
 Example 368
4-(3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
The title compound (22.4 mg, 0.052 mmol, 34.8%) was obtained as white crystals from phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (70 mg, 0.15 mmol) and cyclopropylamine, by the same procedure as in Example 11.
1H-NMR Spectrum (DMSO-d6) δ (ppm): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz).
The starting material was synthesized in the following manner.
Production Example 368-1Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate
The title compound (708 mg, 1.526 mmol, 87.4%) was obtained as light brown crystals from 4-(4-amino-3-chlorophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.745 mmol), by the same procedure as in Production Example 17.
1H-NMR Spectrum (CDCl3) δ (ppm): 4.14 (3H, s), 5.89 (1H, br), 6.50 (1H, d, J=5.6 Hz), 7.16 (2H, dd, J=2.4, 8.8 Hz), 7.22–7.30 (4H, m), 7.44 (2H, m), 7.55 (1H, s), 7.81 (1H, br), 8.31 (1H, d, J=8.8 Hz), 8.68 (1H, d, J=5.6 Hz), 9.27 (1H, s).
Example 368
4-(3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
The title compound (22.4 mg, 0.052 mmol, 34.8%) was obtained as white crystals from phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (70 mg, 0.15 mmol) and cyclopropylamine, by the same procedure as in Example 11.
1H-NMR Spectrum (DMSO-d6) δ (ppm): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz).
The starting material was synthesized in the following manner.
Production Example 368-1Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate
The title compound (708 mg, 1.526 mmol, 87.4%) was obtained as light brown crystals from 4-(4-amino-3-chlorophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.745 mmol), by the same procedure as in Production Example 17.
1H-NMR Spectrum (CDCl3) δ (ppm): 4.14 (3H, s), 5.89 (1H, br), 6.50 (1H, d, J=5.6 Hz), 7.16 (2H, dd, J=2.4, 8.8 Hz), 7.22–7.30 (4H, m), 7.44 (2H, m), 7.55 (1H, s), 7.81 (1H, br), 8.31 (1H, d, J=8.8 Hz), 8.68 (1H, d, J=5.6 Hz), 9.27 (1H, s).........................
CRYSTALLINE FORM
http://www.google.co.in/patents/US7612208 Preparation Example 1 Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (1) Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (17.5 g, 37.7 mmol) disclosed in WO 02/32872 was dissolved in N,N-dimethylformamide (350 mL), and then cyclopropylamine (6.53 mL, 94.25 mmol) was added to the reaction mixture under a nitrogen atmosphere, followed by stirring overnight at room temperature. To the mixture was added water (1.75 L), and the mixture was stirred. Precipitated crude crystals were filtered off, washed with water, and dried at 70° C. for 50 min. To the obtained crude crystals was added ethanol (300 mL), and then the mixture was heated under reflux for 30 min to dissolve, followed by stirring overnight to cool slowly down to room temperature. Precipitated crystals was filtered off and dried under vacuum, and then further dried at 70° C. for 8 hours to give the titled crystals (12.91 g; 80.2%). Preparation Example 2Preparation of 4-(3-cloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (2) (1) Preparation of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

- 1H-NMR Spectrum (CDCl3) δ(ppm): 5.12 (1H, br s), 6.75 (1H, dd, J=9.2, 2.8 Hz), 6.92 (1H, d, J=2.8 Hz), 7.18-7.28 (4H, m), 7.37-7.43 (2H, m), 7.94 (1H, br s). (2) Preparation of 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea

- 1H-NMR Spectrum (CDCl3) δ(ppm): 0.72-0.77 (2H, m), 0.87-0.95 (2H, m), 2.60-2.65 (1H, m), 4.89 (1H, br s), 5.60 (1H, br s), 6.71 (1H, dd, J=8.8, 2.8 Hz), 6.88 (1H, d, J=2.8 Hz), 7.24-7.30 (1H, br s), 7.90 (1H, d, J=8.8 Hz) (3) Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
...................................
PATENT
https://www.google.com/patents/WO2014098176A1?cl=en
According to the present invention 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide amorphous is excellent in solubility in water. Example 1 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide manufacture of amorphous amide 4- (3-chloro-4- (cyclopropylamino-carbonyl) amino phenoxy) -7-methoxy-6-quinolinecarboxamide B-type crystals (Patent Document 2) were weighed to 300mg, is placed in a beaker of 200mL volume, it was added tert- butyl alcohol (tBA) 40mL. This was heated to boiling on a hot plate, an appropriate amount of tBA to Compound A is dissolved, water was added 10mL. Then, the weakened heated to the extent that the solution does not boil, to obtain a sample solution. It should be noted, finally the solvent amount I was 60mL. 200mL capacity eggplant type flask (egg-plant shaped flask), and rotated in a state of being immersed in ethanol which had been cooled with dry ice. It was added dropwise a sample solution into the interior of the flask and frozen. After freezing the sample solution total volume, to cover the opening of the flask in wiping cloth, and freeze-dried. We got an amorphous A of 290mg.
Patent Document 2: US Patent Application Publication No. 2007/0117842 Patent specification

Amorphous A 13 C-solid state NMR spectrum in Figure 2, the chemical shifts and I are shown in Table 3.
[Table 3] *: peak of t- butyl alcohol

.............................
Paper
ACS Medicinal Chemistry Letters (2015), 6(1), 89-94 http://pubs.acs.org/doi/full/10.1021/ml500394m
.................
Paper
Journal of Pharmaceutical and Biomedical Analysis (2015), 114, 82-87 http://www.sciencedirect.com/science/article/pii/S0731708515002940
KEEP WATCHING WILL BE UPDATED.............most of my posts are updated regularly
References
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. (2008). "Multi-Kinase Inhibitor E7080 Suppresses Lymph Node and Lung Metastases of Human Mammary Breast Tumor MDA-MB-231 via Inhibition of Vascular Endothelial Growth Factor-Receptor (VEGF-R) 2 and VEGF-R3 Kinase". Clinical Cancer Research 14 (17): 5459–65.doi:10.1158/1078-0432.CCR-07-5270. PMID 18765537.
- "Phae III trial shows lenvatinib meets primary endpoint of progression free surival benefit in treatment of radioiodine-refactory differentiated thyroid cancer". Eisai. 3 February 2014.
- Glen, H; D. Boss; T. R. Evans; M. Roelvink; J. M. Saro; P. Bezodis; W. Copalu; A. Das; G. Crosswell; J. H. Schellens (2007). "A phase I dose finding study of E7080 in patients (pts) with advanced malignancies". Journal of Clinical Oncology, ASCO Annual Meeting Proceedings Part I 25 (18S): 14073.
- ClinicalTrials.gov NCT00946153 Study of E7080 in Patients With Advanced Hepatocellular Carcinoma (HCC)
- Gild, M. L.; Bullock, M.; Robinson, B. G.; Clifton-Bligh, R. (2011). "Multikinase inhibitors: A new option for the treatment of thyroid cancer". Nature Reviews Endocrinology 7 (10): 617–624.doi:10.1038/nrendo.2011.141. PMID 21862995.
- ClinicalTrials.gov NCT01321554 A Trial of E7080 in 131I-Refractory Differentiated Thyroid Cancer
EXTRAS
...............
Martin Schlumberger et al. A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib(E7080) in patients with 131I-refractory differentiated thyroid cancer (SELECT). 2014 ASCO Annual Meeting. Abstract Number:LBA6008. Presented June 2, 2014. Citation: J Clin Oncol 32:5s, 2014 (suppl; abstr LBA6008). Clinical trial information: NCT01321554. Bando, Masashi. Quinoline derivative-containing pharmaceutical composition. PCT Int. Appl. (2011), WO 2011021597 A1 Tomohiro Matsushima, four Nakamura, Kazuhiro Murakami, Atsushi Hoteido, Yusuke Ayat, Naoko Suzuki, Itaru Arimoto, Pinche Hirose, Masaharu Gotoda.Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor. US patent number US7612208 Also published as: CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1 , US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date: Aug 7, 2007 Original Assignee: Eisai Co., Ltd Funahashi, Yasuhiro et al.Preparation of urea derivatives containing nitrogenous aromatic ring compounds as inhibitors of angiogenesis. US patent number US7253286, Also published as:CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1, US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date:Aug 7, 2007. Original Assignee:Eisai Co., Ltd Sakaguchi, Takahisa; Tsuruoka, Akihiko. Preparation of amorphous salts of 4-[3-chloro-4-[(cyclopropylaminocarbonyl)amino]phenoxy]-7-methoxy-6-quinolinecarboxamide as antitumor agents. PCT Int. Appl. (2006), WO2006137474 A1 20061228. Naito, Toshihiko and Yoshizawa, Kazuhiro. Preparation of urea moiety-containing quinolinecarboxamide derivatives. PCT Int. Appl., WO2005044788, 19 May 2005 Itaru Arimoto et al. Crystal of salt of 4-[3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy]-7-methoxy-6-quinolinecarboxamide or solvate thereof and processes for producing these. PCT Int. Appl. (2005), WO2005063713 A1 20050714.
10-23-2009
|
ANTITUMOR AGENT FOR UNDIFFERENTIATED GASTRIC CANCER
|
|
10-2-2009
|
ANTI-TUMOR AGENT FOR MULTIPLE MYELOMA
|
|
8-21-2009
|
ANTITUMOR AGENT FOR THYROID CANCER
|
|
8-14-2009
|
THERAPEUTIC AGENT FOR LIVER FIBROSIS
|
|
2-27-2009
|
USE OF COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND c-kit KINASE INHIBITOR
|
|
9-5-2008
|
Medicinal Composition
|
|
8-8-2007
|
Nitrogen-containing aromatic derivatives
|
|
5-25-2007
|
Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same
|
|
7-21-2006
|
Nitrogen-containing aromatic derivatives
|
|
6-23-2006
|
Use of sulfonamide-including compounds in combination with angiogenesis inhibitors
|
11-16-2011
|
UREA DERIVATIVE AND PROCESS FOR PREPARING THE SAME
|
|
8-10-2011
|
c-Kit kinase inhibitor
|
|
7-6-2011
|
Nitrogen-Containing Aromatic Derivatives
|
|
12-24-2010
|
COMBINED USE OF ANGIOGENESIS INHIBITOR AND TAXANE
|
|
9-24-2010
|
COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND ANTI-TUMOR PLATINUM COMPLEX
|
|
4-30-2010
|
METHOD FOR PREDICTION OF THE EFFICACY OF VASCULARIZATION INHIBITOR
|
|
4-16-2010
|
METHOD FOR ASSAY ON THE EFFECT OF VASCULARIZATION INHIBITOR
|
|
3-24-2010
|
Urea derivative and process for preparing the same
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF PANCREATIC CANCER
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF UNDIFFERENTIATED GASTRIC CANCER
|
| US7253286 * | 18 Apr 2003 | 7 Aug 2007 | Eisai Co., Ltd | Nitrogen-containing aromatic derivatives |
| US20040053908 | 18 Apr 2003 | 18 Mar 2004 | Yasuhiro Funahashi | Nitrogen-containing aromatic derivatives |
| US20040242506 | 9 Aug 2002 | 2 Dec 2004 | Barges Causeret Nathalie Claude Marianne | Formed from paroxetine hydrochloride and ammonium glycyrrhyzinate by precipitation, spray, vacuum or freeze drying, or evaporation to glass; solid or oil; masks the bitter taste of paroxetine and has a distinctive licorice flavor; antidepressants; Parkinson's disease |
| US20040253205 | 10 Mar 2004 | 16 Dec 2004 | Yuji Yamamoto | c-Kit kinase inhibitor |
| US20070004773 * | 22 Jun 2006 | 4 Jan 2007 | Eisai R&D Management Co., Ltd. | Amorphous salt of 4-(3-chiloro-4-(cycloproplylaminocarbonyl)aminophenoxy)-7-method-6-quinolinecarboxamide and process for preparing the same |
| US20070078159 | 22 Dec 2004 | 5 Apr 2007 | Tomohiro Matsushima | Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor |
| US20070117842 * | 22 Apr 2004 | 24 May 2007 | Itaru Arimoto | Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same |
| EP0297580A1 | 30 Jun 1988 | 4 Jan 1989 | E.R. SQUIBB & SONS, INC. | Amorphous form of aztreonam |
| JP2001131071A | Title not available | |||
| JP2005501074A | Title not available | |||
| JPS6422874U | Title not available | |||
| WO2002032872A1 | 19 Oct 2001 | 25 Apr 2002 | Itaru Arimoto | Nitrogenous aromatic ring compounds |
| WO2003013529A1 | 9 Aug 2002 | 20 Feb 2003 | Barges Causeret Nathalie Claud | Paroxetine glycyrrhizinate |
| WO2004039782A1 | 29 Oct 2003 | 13 May 2004 | Hirai Naoko | QUINOLINE DERIVATIVES AND QUINAZOLINE DERIVATIVES INHIBITING AUTOPHOSPHORYLATION OF Flt3 AND MEDICINAL COMPOSITIONS CONTAINING THE SAME |
| WO2004080462A1 | 10 Mar 2004 | 23 Sep 2004 | Eisai Co Ltd | c-Kit KINASE INHIBITOR |
| WO2004101526A1 | 22 Apr 2004 | 25 Nov 2004 | Itaru Arimoto | Polymorphous crystal of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-qunolinecarboxamide and method for preparation thereof |
| WO2005044788A1 | 8 Nov 2004 | 19 May 2005 | Eisai Co Ltd | Urea derivative and process for producing the same |
| WO2005063713A1 | 22 Dec 2004 | 14 Jul 2005 | Itaru Arimoto | Crystal of salt of 4-(3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy)-7-methoxy-6-quinolinecarboxamide or of solvate thereof and processes for producing these |
| WO2006030826A1 | 14 Sep 2005 | 23 Mar 2006 | Eisai Co Ltd | Medicinal composition |
1H NMR PREDICT OF LENVATINIB BASE


सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL


















 7-Chloro-5-oxo-2,3,4,5-tetrahydro-lH-l
-benzazepine
7-Chloro-5-oxo-2,3,4,5-tetrahydro-lH-l
-benzazepine


 2-methyl benzoyl chloride
2-methyl benzoyl chloride




