RP 6503
phase 1

RP 6503
| Molecular Formula: | C30H24F2N6O5S |
|---|---|
| Molecular Weight: | 618.610566 g/mol |

A1
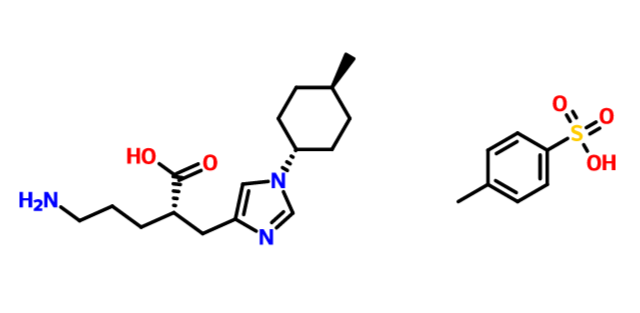
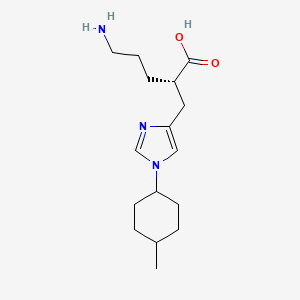
RP 6503
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl) ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
(S)-N-[5-[4-amino-1-[1-[5-fluoro-3-(3-fluorophenyl)-4-oxochromen-2-yl]ethyl]pyrazolo[3,4-d]pyrimidin-3-yl]-2-methoxyphenyl]methanesulfonamide


Novartis to develop and commercialize Rhizen's inhaled dual PI3K-delta gamma inhibitor and related compounds worldwide
The immune pipeline includes ‘dual PI3K inhibitors for various indications’ licensed to Novartis
‘inhaled dual inhibitor’,
Phosphoinositide-3 kinase delta inhibitor; Phosphoinositide-3 kinase gamma inhibitor
WO2011055215A2 and WO2012151525A1 and U.S. Publication Nos. US20110118257 and US20120289496
Rhizen Pharmaceuticals Sa INNOVATOR
| Incozen Therapeutics Pvt. Ltd., Rhizen Pharmaceuticals Sa |
http://www.google.com/patents/WO2011055215A2?cl=en
PATENT
http://www.google.com/patents/WO2012151525A1?cl=en
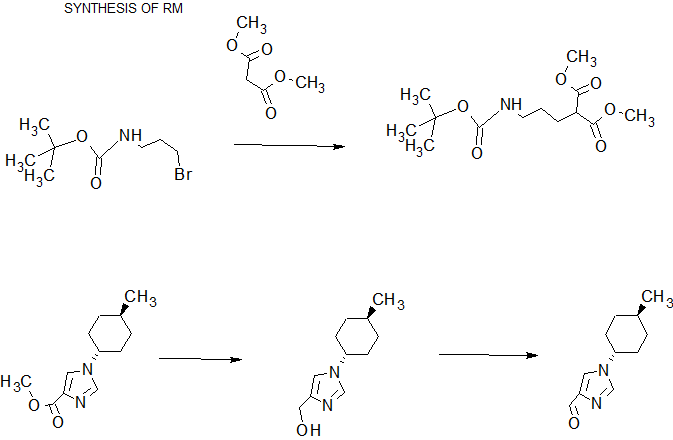
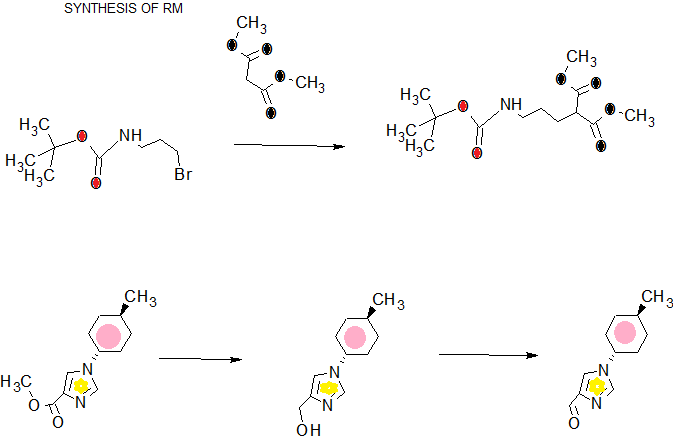
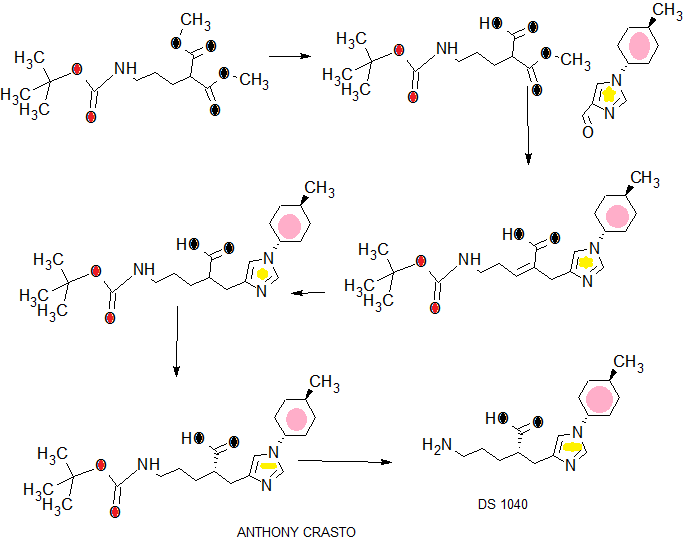
scheme 1A:
Ste -1



SCHEME 3






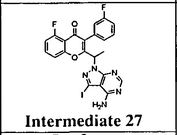
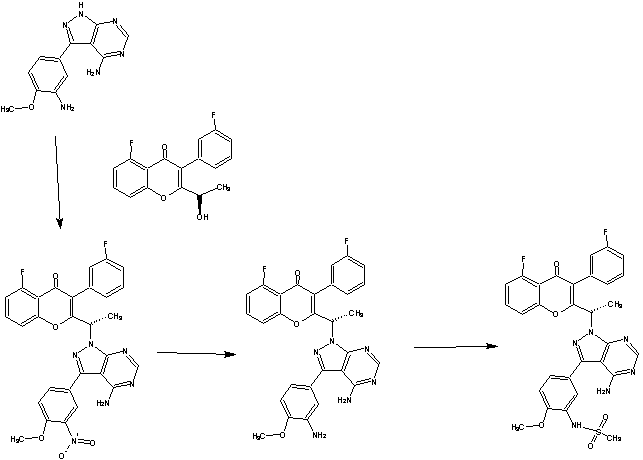
Intermediate 27: 2-( l -(4-amino-3-iodo-lH-pyrazolo[3,4-d]pyrimidin- l - yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: To a solution of 3-iodo- l H- pyrazolo[3,4-d]pyrimidin-4-amine (0.800 g, 2.88 mmol) in DMF (5 ml), potassium carbonate (0.398 g, 2.88 mmol) was added and stirred at RT for 30 min. To this mixture intermediate 22 (0.500 g, 1.44 mmol) was added and stirred for 12h. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a off-white solid (0.300 g, 38%). Ή-NMR (5 ppm, DMSO-d63, 400 MHz): 8.02 (s, 1 H), 7.94 (s, 1 H), 7.84 (dt, J = 8.4,5.7 Hz, 1H), 7.47 (d, 7 = 8.6 Hz, 1H), 7.29 (m, 3H), 7.09 (dt, 7 = 8.8,2.3 Hz, 1 H), 6.87 (s, 2H), 5.88 (q, 7 = 7.0 Hz, 1H), 1.82 (d, 7 = 7.0 Hz, 3H).
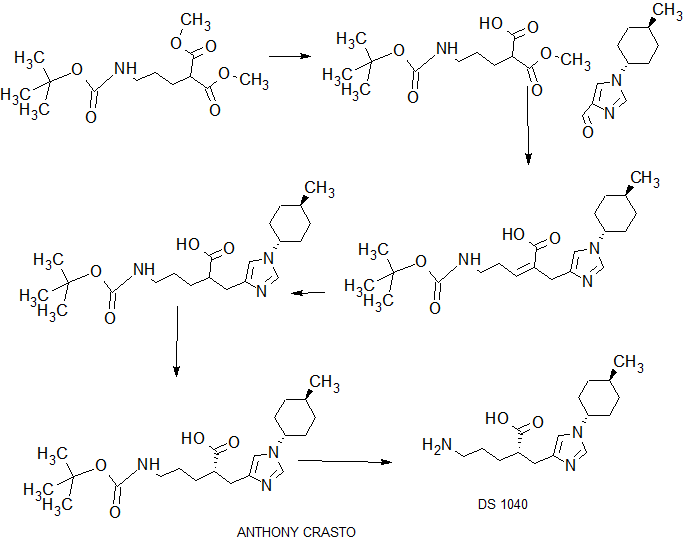
SYNTHESIS
MAIN PART
PATENT
http://www.google.com/patents/WO2015198289A1?cl=enPrashant Kashinath Bhavar, Swaroop Kumar Venkata Satya VAKKALANKA
The present invention relates to a selective dual delta (δ) and gamma (γ) PI3K protein kinase modulator (S)-N-(5-(4-amino-1-(1-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H- chromen-2-yl)ethyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl) methane sulfonamide, methods of preparing them, pharmaceutical compositions containing them and methods of treatment, prevention and/or amelioration of PI3K kinase mediated diseases or disorders with them.
compound of formula (Al):
(Al).
The process comprises the steps of:
(a) subjecting (R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one:

to a Mitsunobu reaction with 3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-amine:

(for example, in the presence of triphenylphosphine and diisopropylazodicarboxylate) to give (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (Intermediate 3):

Intermediate 3;
(b) reducing Intermediate 3, for example with a reducing agent such as Raney Ni, to give (S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin- l-yl)ethyl)-5-fluoro-3-( -fluorophenyl)-4H-chromen-4-one (Intermediate 4):

Intermediate 4;
The intermediates described herein may be prepared by the methods described in International Publication Nos. WO 11/055215 and WO 12/151525, both of which are hereby incorporated by reference.
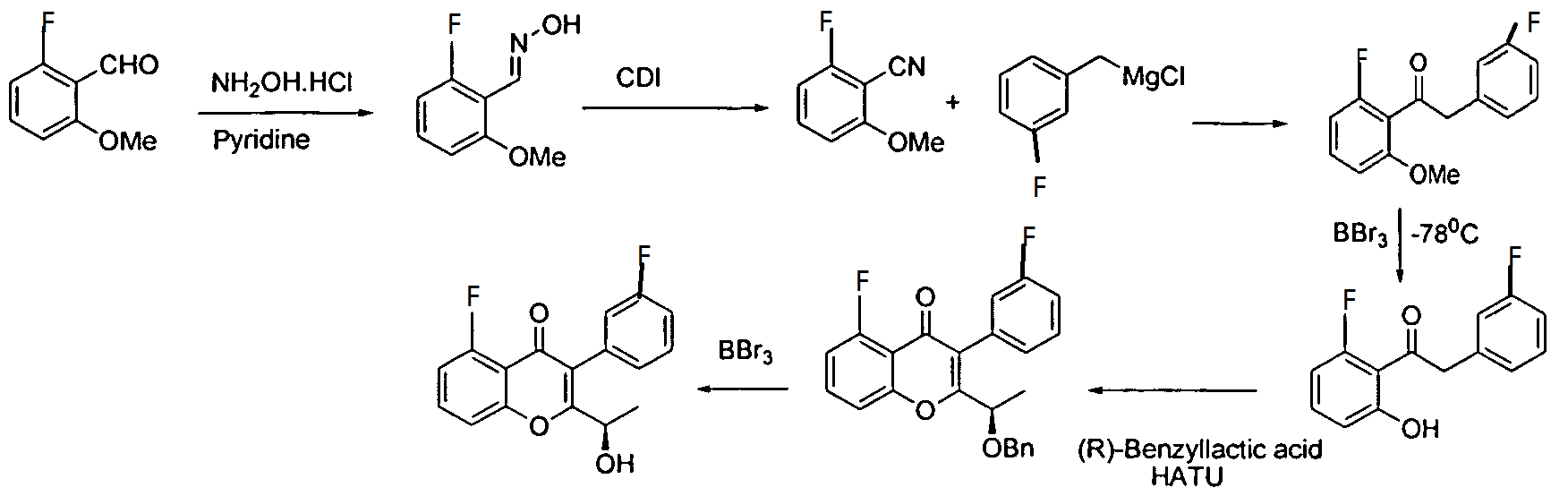
Intermediate 1: N-(5-bromo-2-methoxyphenyl)methanesulfonamide:
To a solution of 5-bromo-2-methoxyaniline(1.00 g, 4.94 mmol) in dichloromethane (10 ml), pyridine (0.800 ml, 9.89 mmol) was added and cooled to 0°C. Methane sulphonyl chloride (0.40 ml, 5.19 mmol) was added and stirred for 30 min. The reaction mixture was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was chromatographed with ethyl acetate : petroleum ether to afford the title compound as a reddish solid (1.20 g, 87%).
Intermediate 2: N-(2-methoxy-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)methanesulfonamide: Potassium acetate (0.841 g, 8.57 mmol) and bis(pinacolato)diboron (1.190 g, 4.71 mmol) were added to a solution of intermediate 1 (1.20 g, 4.28 mmol) in dioxane (17.5 ml) and the solution was degassed for 30 min.[l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Ci2 (0.104 g, 0.128 mmol) was added under nitrogen atmosphere and heated to 80°C. After 2h the
reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as a yellow solid (1.00 g, 71%).JH-NMR (δ ppm, CDCb, 400 MHz): 7. 91 (d, / = 1.2Hz, 1H), 7. 62 (dd, / = 8.1, 1.2Hz, 1H), 6. 92 (d, / = 8.1Hz, 1H), 6.73 (s, 1H), 3.91 (s, 3H), 2.98 (s, 3H), 1.32 (s, 12H).
Intermediate 3: (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: To a solution of (R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one (0.500 g, 1.64 mmol) in THF (5 ml), 3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-amine (0.564 g, 1.97 mmol) and triphenylphosphine (0.649 g, 2.47 mmol) were added followed by the addition of diisopropylazodicarboxylate (0.50 ml, 2.47 mmol). ((R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one can be prepared as described for Intermediates 23, 25, and 26 in International Publication No. WO 2012/0151525.). After 4h at room temperature, the mixture was concentrated and the residue was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as a brown solid (0.270 g, 29%). JH-NMR (δ ppm, DMSO-d6, 400 MHz): 8.04 (s, 1H), 7.83 (m, 1H), 7.63-7.50 (m, 3H), 7.29 (m, 2H), 7.06 (dt, J = 8.7,2.2Hz, 1H), 6.94 (m, 2H), 6.75 (dd, J = 8.1,2.1Hz, 1H), 5.95 (q, J = 7.0Hz, 1H), 4.98 (s, 2H), 3.81 (s, 3H), 1.86 (d, J = 7.0 Hz, 3H).
[109] Intermediate 4: (S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one:
(S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one : To a solution of Intermediate 3 (0.260 g, 0.455 mmol) in ethanol (5 ml), Raney Ni (0.130 g) was added and hydrogeneated at 20psi at 50°C for 24h. The reaction mixture was passed through celitepad and concentrated to afford the title compound as a brown solid (0.150 g, 60%). Mass : 540.8 (M+).
Example A
N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH- pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
To a solution of 2-(l-(4-amino-3-iodo-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (0.200 g, 0.366 mmol) in DME (2.1 ml) and water (0.67 ml), intermediate 2 (0.179 g, 0.550 mmol) and sodium carbonate (0.116 g, 1.10 mmol) were added and the system was degassed for 30 min. (2-(l-(4-amino-3-iodo-lH^yrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one can be prepared as described for Intermediates 23, 25, and 26 in International Publication No. WO 2012/0151525). Bis(diphenylphosphino) ferrocene]dichloropalladium(II) (0.059 g, 0.075 mmol) was added and kept under microwave irradiation (microwave power = 100W, temperature = 100 °C) for 45 min. The reaction mixture was Celite filtered, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a brown solid (0.080 g, 35%). MP: 216-218 °C. ¾-NMR (δ ppm, CDCb, 400 MHz): 8.20 (s, 1H), 7.73 (s, 1H), 7.53 (m, 2H), 7.31 (m, 2H), 7.07-6.73 (m, 6H), 6.07 (q, / = 6.2 Hz, 1H), 3.98 (s, 3H), 3.14 (s, 3H), 2.01 (d, / = 6.0Hz, 3H).
Example Al and A2
Method A
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
and (R)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2- yl)ethyl)-lH-p anesulfonamide

The two enantiomerically pure isomers were separated by preparative SFC (supercritical fluid) conditions from N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide (0.500 g) on a CHIRALPAK AS-H column (250 x 30 mm; 5μπι) using methanol : CO2 (55:45) as the mobile phase at a flow rate of 80g / min.
Example Al (S-isomer): Brown solid (0.247 g). Enantiomeric excess: 97.4%. Retention time: 2.14 min. Mass: 619.1 (M++l). MP: 156-158° C.
Example A2 (R-isomer): Brown solid (0.182 g). Enantiomeric excess: 99.3%. Retention t: 3.43 min. Mass: 619.1 (M++l). MP: 168-171° C.
Method Al
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
and (R)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2- yl)ethyl)-lH-p anesulfonamide

The two enantiomerically pure isomers were separated by preparative SFC (supercritical fluid) conditions from N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl) methanesulfonamide (15.0 g) on a CHIRALPAK AS-H column (250 x 20 mm; 5μπι) using methanol : CO2 (45:55) as the mobile phase at a flow rate of 120g / min.
Example Al (S-isomer): Enantiomeric excess: 100 %. Retention time: 2.21 min. Mass: 619.1 (M++l). MP: 175-178° C Specific optical rotation (C=l in chloroform, at 25°C) : [a]D = + 147.16.
Example A2 (R-isomer): Enantiomeric excess: 99.3%. Retention t: 3.72 min. Mass: 619.1 (M++l). MP: 154-157° C. Specific optical rotation (C=l in chloroform, at 25°C) : [a]D = - 159.54.
Method B
Example Al
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
To a solution of Intermediate 4 (0.500 g, 0.923 mmol) in dichloromethane (5 ml) cooled to 0°C, pyridine (0.200 ml, 1.84 mmol) was added and stirred for 10 min. Methanesulphonyl chloride (0.100 ml, 0.923 mmol) was added stirred for 30 min. The reaction mixture was quenched with water, extracted with dichloromethane and dried over sodium sulphate. The crude product was column chromatographed with methanol : dichloromethane to afford the title compound as an off-white solid (0.240 g, 42%). MP: 211-213°C. ¾-NMR (δ ppm, DMSO-d6, 400 MHz): 9.15 (s, 1H), 8.06 (s, 1H), 7.83 (m, 1H), 7.49 (m, 4H), 7.28 (m, 4H), 7.08 (dt, / = 8.6, 1.7 Hz, 1H), 6.92 (s, 2H), 5.98 (q, / = 6.9 Hz, 1H), 3.88 (s, 3H), 2.99 (s, 3H), 1.88 (d, / = 7.0 Hz, 3H). Enantiomeric excess: 85.4% as determined by HPLC on a chiralpak AS-3R column, enriched in the fast eluting isomer (retention time = 7.46 min.).

CLIPS
La Chaux-de-Fonds, Switzerland, Sept. 6, 2013 -- La Chaux-de-Fonds, Switzerland (6 September 2013): Rhizen Pharmaceuticals S.A. announces a scientific poster presentation on the pre-clinical characterization of its lead calcium release activated channel (CRAC) inhibitor, RP3128, for the treatment of respiratory disorders and an oral presentation on the pharmacological profile of its novel, dual Phosphoinositide-3 kinase (PI3K) delta/gamma inhibitor, RP6503, in the pulmonary disease systems, at the European Respiratory Society Annual Congress (ERS), to be held from 7-11 September 2013, at Barcelona, Spain.
RP6503 is a novel, potent and selective inhibitor of the delta and gamma isoforms of PI3K. It is to be delivered via the inhalation route and has a long duration of action along with excellent PI3K isoform selectivity, which is expected to result in better safety. RP3128 has been optimized with high potency for CRAC channel inhibition, selectivity over the other voltage gated channels and excellent oral bioavailability. Rhizen intends to move both these compounds to the clinic in 2014.
Details of the presentations:
1. Abstract of the Poster Presentation: "Pre-clinical characterization of RP3128, a novel and potent CRAC channel inhibitor for the treatment of respiratory disorders"
Time and Location- 8 September 2013 between 14.45-16.45 in Room 3.6, at Poster Discussion: New drugs in respiratory medicine, at FIRA BARCELONA, Convention Centre de Gran Via, Barcelona, Spain
2. Abstract of Oral Presentation: "In vitro and in vivo pharmacological profile of RP6503, a novel dual PI3K delta/gamma inhibitor, in pulmonary disease systems"
Time and Location- 11 September 2013 at 8.45 in Room 3.9; Session 8.30-10.30, at the Oral Presentation: Emerging new targets for the treatment of respiratory diseases, at FIRA BARCELONA, Convention Centre de Gran Via, Barcelona, Spain
CLIPS
La Chaux-de-Fonds, Switzerland , Dec. 09, 2015 -- Rhizen Pharmaceuticals S.A. announced today that they have entered into an exclusive, worldwide license agreement with Novartis for the development and commercialization of Rhizen's, inhaled dual PI3K-delta gamma inhibitor and its closely related compounds for various indications.
Under the terms of the agreement, Rhizen will receive an upfront payment and is eligible to receive development, regulatory and sales milestones payments. In addition Rhizen is also eligible to receive tiered royalties on annual nets sales.
The lead compound is a novel, potent, and selective dual PI3K-delta gamma inhibitor with demonstrated anti-inflammatory and immuno-modulatory activity in pre-clinical systems and models representative of respiratory diseases. With a favorable ADME and PK profile and high therapeutic index in animals, the inhaled dual PI3K-delta gamma inhibitor holds promise in the treatment of human airway disorders.
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen's website, www.rhizen.com.
SEE
https://newdrugapprovals.org/2015/12/10/alembic-pharma-advances-1-on-rhizen-novartis-license-agreement/
WO-2015181728
WO-2015001491
WO-2014072937
WO-2014006572
http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A3880
| WO2011055215A2 | Nov 3, 2010 | May 12, 2011 | Incozen Therapeutics Pvt. Ltd. | Novel kinase modulators |
| WO2012008302A1 | Jun 28, 2011 | Jan 19, 2012 | National University Corporation Tottori University | Method for preparing novel hipsc by means of mirna introduction |
| WO2012121953A1 | Feb 29, 2012 | Sep 13, 2012 | The Trustees Of Columbia University In The City Of New York | Methods and pharmaceutical compositions for treating lymphoid malignancy |
| WO2012151525A1 | May 4, 2012 | Nov 8, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
| WO2013164801A1 | May 3, 2013 | Nov 7, 2013 | Rhizen Pharmaceuticals Sa | Process for preparation of optically pure and optionally substituted 2- (1 -hydroxy- alkyl) - chromen - 4 - one derivatives and their use in preparing pharmaceuticals |
| US20110118257 | May 19, 2011 | Rhizen Pharmaceuticals Sa | Novel kinase modulators | |
| US20120289496 | May 4, 2012 | Nov 15, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
///////RP 6503, Novartis, develop, commercialize, Rhizen, inhaled dual PI3K-delta gamma inhibitor, PHASE 1, RP-6503
c21c(cccc1O/C(=C(\C2=O)c3cc(ccc3)F)C(C)n4c6ncnc(c6c(n4)c5cc(c(cc5)OC)NS(=O)(=O)C)N)F
CC(C1=C(C(=O)C2=C(O1)C=CC=C2F)C3=CC(=CC=C3)F)N4C5=C(C(=N4)C6=CC(=C(C=C6)OC)NS(=O)(=O)C)C(=NC=N5)N

/////