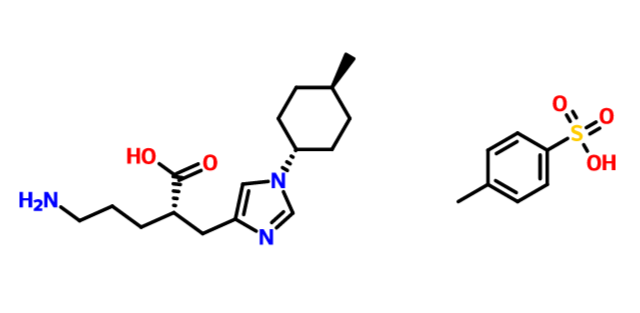
DS-1040
Daiichi Sankyo Co Ltd
Ischemic stroke
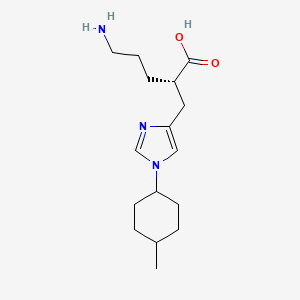
(2S)-5-amino-2-[[1-(4-methylcyclohexyl)imidazol-4-yl]methyl]pentanoic acid
1H-Imidazole-4-propanoic acid, α-(3-aminopropyl)-1-(trans-4-methylcyclohexyl)-, (αS)-
(2S)-5-amino-2-{[1-(trans-4-methylcyclohexyl)-1H-imidazol-4-yl]methyl}pentanoic acid
free form cas 1335138-62-9
1:1 TOSYLATE 1335138-89-0
1335138-90-3 1:1:1 TOSYLATE HYDRATE
phase 2, Ischemic stroke
| Molecular Formula: | C16H27N3O2 |
|---|---|
| Molecular Weight: | 293.40448 g/mol |

TAFIa inhibitors, useful for treating myocardial infarction, angina, pulmonary hypertension and deep vein thrombosis.
In March 2016, DS-1040 was reported to be in phase 2 C clinical development, and the study was expected to complete in June 2017.
https://clinicaltrials.gov/ct2/show/NCT02560688
- 01 Feb 2016Daiichi Sankyo initiates a phase I trial in Healthy volunteers in United Kingdom (NCT02647307)
- 09 Jan 2016Daiichi Sankyo plans a phase I trial in Healthy volunteers in United Kingdom (NCT02647307)
- 29 Sep 2015Daiichi Sankyo plans a drug-interaction phase I trial in Healthy volunteers in United Kingdom (IV) (NCT02560688)
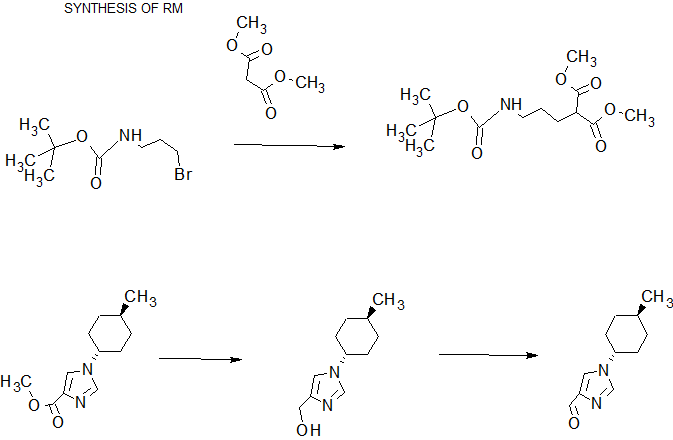
SYNTHESIS
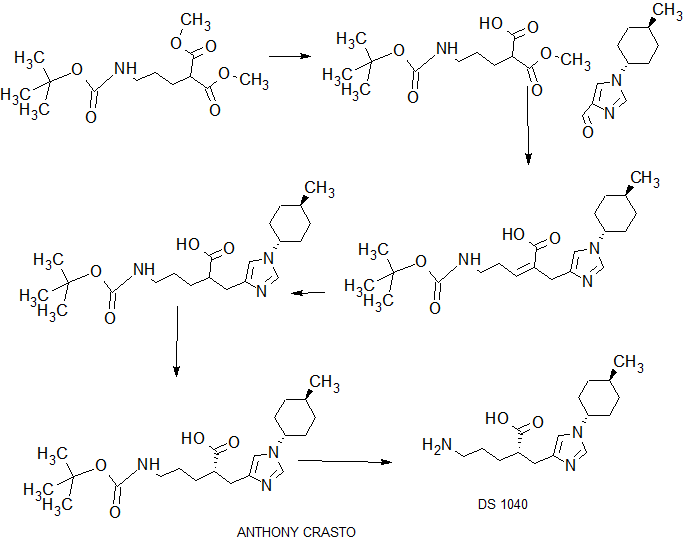
COMPLETE SYNTHESIS
Patent
WO201111506
PATENT
WO2013039202
PATENT
WO 2016043254
PATENT
WO2016043253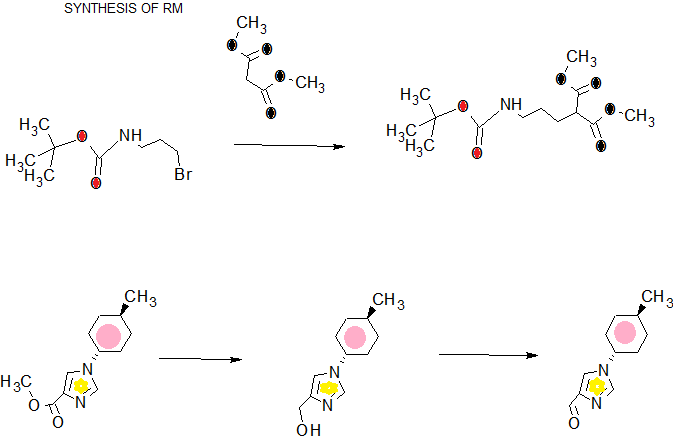
COMPLETE SYN..........
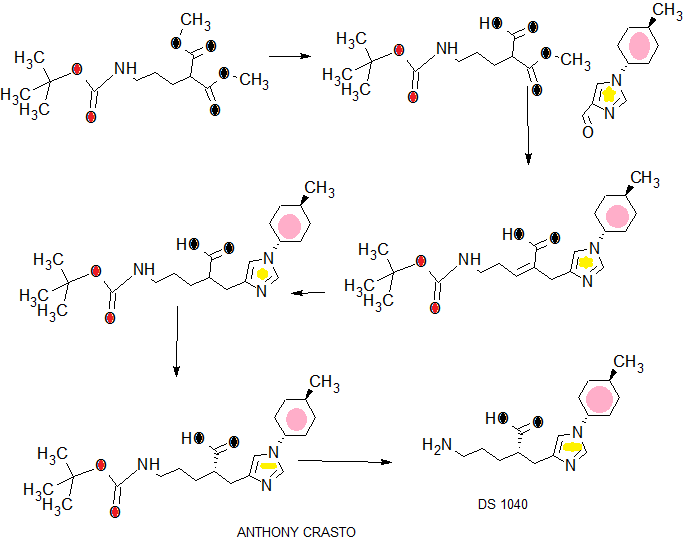
WO2016043253
The optical purity of the obtained compound was measured by the following HPLC analysis conditions.
(2S) -5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans -4- methylcyclohexyl)-lH-imidazol-4-yl] methyl} valeric acid (S)-2-amino 1-propanol salt (A1 step, A2 step, A3 step), (2S) -5 - [ (tert- butoxycarbonyl) amino] -2 - {[1- (trans -4- methylcyclohexyl)-lH-imidazole 4-yl] methyl} optical purity measurement conditions valerate (A4 step):
column: CHIRAL AGP 4.6mmI. D. × 250mm (5μm),
mobile phase: methanol / 10mM phosphate buffer solution (pH7.0) = 95/5,
temperature: 40 ℃,
flow rate: 0.5mL / min,
detection method: UV at 220nm,
retention time: R body: 5.9 minutes, S body: 7.3 minutes.
(2S) -5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans -4- methylcyclohexyl)-lH-imidazol-4-yl] methyl} valeric acid (S)-2-amino 1-propanol salt (A1 step, A2 step, A3 step), (2S) -5 - [ (tert- butoxycarbonyl) amino] -2 - {[1- (trans -4- methylcyclohexyl)-lH-imidazole 4-yl] methyl} optical purity measurement conditions valerate (A4 step):
column: CHIRAL AGP 4.6mmI. D. × 250mm (5μm),
mobile phase: methanol / 10mM phosphate buffer solution (pH7.0) = 95/5,
temperature: 40 ℃,
flow rate: 0.5mL / min,
detection method: UV at 220nm,
retention time: R body: 5.9 minutes, S body: 7.3 minutes.
column: CHIRLCEL OZ-H 4.6mmI. D. × 250mm (5μm),
mobile phase: hexane / ethanol / methanol / isopropanol / trifluoroacetic acid / triethylamine = 860/100/20/2/2
temperature: 30 ℃
flow rate: 1.0mL / min
detection method: UV at 220nm
retention time: R body: 16.1 minutes, S body: 13.0 minutes (example 1) (1-1) 5 - [(Tert- butoxycarbonyl) amino] -2-methoxy-carbonyl) valeric acid morpholine salt
[Of 11]

In
methanol (400mL) solution of di -tert- butyl (100.0g) and
3-chloro-propylamine hydrochloride (71.5g), was added dropwise
triethylamine (51.0g) at 0 ℃, at the same temperature It was stirred for
16 hours. To the reaction solution was added toluene (400 mL) and water
(400 mL), then were separated, and the organic layer was washed with
water. Toluene 400mL was added to the organic layer, was concentrated
under reduced pressure to 300 mL, N, N-dimethylacetamide (210 mL) was
added and concentrated in vacuo to 300 mL. Potassium carbonate solution
(126.66g), tetrabutylammonium bromide (44.32g), was added dimethyl
malonate (90.82g) and N, N-dimethylacetamide (100 mL), stirred for 20
hours at 55 ° C. did. Toluene (400 mL) and water (700 mL) was added to
the reaction mixture, after separation, The organic layer was washed
with water, with 1M aqueous sodium hydroxide and water, and concentrated
under reduced pressure to 150 mL. This solution methanol (1870mL) and
1M sodium hydroxide solution (430.8mL) in addition to, and the mixture
was stirred for 27.5 hours at 0 ℃. Concentrated hydrochloric acid to the
reaction solution (2.5 mL) was added, the pH was adjusted to 7-9, and
concentrated in vacuo to 375 mL. After addition of ethyl acetate (500mL)
to the reaction solution, concentrated hydrochloric acid (35.1mL) was
added, the pH was adjusted to 2.2-2.5, and the layers were separated.
The aqueous layer was extracted with ethyl acetate (500 mL), after
mixing the organic layer under reduced pressure, and prepared by
dehydration condensation of ethyl acetate (250 mL) solution. The
resulting solution of ethyl acetate (500 mL) and morpholine (37.5 g) was
added to and stirred overnight. The precipitated crystals were
filtered, washed with ethyl acetate, and dried under reduced pressure,
to give the title compound (136.1g, 81.9% yield).
(1-2) [1- (trans-4- methylcyclohexyl) -1H- imidazole-4-yl] methanol
[Of 12]

N,
and stirred for 4 h methanol (56 mL) solution at 5 ~ 10 ℃ of N-
dimethylformamide dimethyl acetal (77.4 g) and ethyl isocyanoacetate
(70.0g).The reaction solution was cooled to 0 ℃, water (5.3mL) and
trans-4- methylcyclohexyl amine (105.1g) was added, and the mixture was
stirred for 24 hours at 60 ~ 65 ℃. The reaction was cooled to room
temperature, toluene (420 mL), supplemented with 10% brine (280 mL) and
concentrated hydrochloric acid (68 mL), After separation, the organic
layer was washed with 10% brine (140 mL). Organic layer to 10% sodium
chloride solution (280mL) and concentrated hydrochloric acid were added
for liquid separation after (78.4g), was added to separate liquid
further 10% saline solution into the organic layer (210mL) and
concentrated hydrochloric acid (31.3g). After dissolving sodium chloride
(70.0 g) in aqueous layer, adding toluene (420 mL) and 50% aqueous
sodium hydroxide (85 mL), after separation, toluene (350 mL) the organic
layer was added, under reduced pressure, dehydration concentrated was
prepared in toluene (420 mL) solution was. The solution was cooled to 0
℃, dropped the hydrogenated bis (2-methoxyethoxy) aluminum sodium (70%
toluene solution) (207.4g), and the mixture was stirred at room
temperature for 1 hour. The reaction was cooled to 0 ° C., was added
dropwise 12.5% aqueous sodium hydroxide solution (700 mL), stirred for
1 hour at room temperature. After the solution was separated and the
organic layer was washed successively with 12.5% aqueous solution of
sodium hydroxide (700mL) and 20% sodium chloride solution (140mL),
toluene in the organic layer (140mL), 1- butanol (14mL), water ( 280mL)
and was added to aliquots of concentrated hydrochloric acid (48mL). It
was further added to liquid separation with water (140 mL) and
concentrated hydrochloric acid (2 mL) to the organic layer. Met The
aqueous layer was stirred in for 1 hour activated carbon (10.5 g),
activated charcoal was filtered off, the activated carbon was washed
with water (210 mL). Matches the filtrate and washings, sodium chloride
(140 g), toluene was added (980 mL) and 50% aqueous sodium hydroxide (42
mL), After separation, under reduced pressure and the organic layer was
dried concentrated toluene (210 mL) It was prepared in solution. The
solution was stirred 30 minutes at 50-55 ° C., cooled to room
temperature, it was added dropwise heptane (560 mL), and stirred at the
same temperature for 3 hours. The precipitated crystals were filtered to
give after washing with toluene / heptane (1/4) mixture solution, the
title compound was dried under reduced pressure (77.2 g, 64.2% yield).
1 H-NMR (CDCl 3
) [delta]: 7.49 (1H, s), 6.91 (1H, s), 4.58 (2H, s), 3.83 (1H, tt, J =
12.0 , 3.9Hz), 2.10-2.07 (2H, m), 1.87-1.84 (2H, m), 1.70-1.61 (2H, m),
1.48-1 .42 (1H, m), 1.15-1.06 (2H, m), 0.95 (3H, d, J = 6.5Hz).
[Of 13]

(1-2)
The compound obtained in (50.0 g) in toluene (350 mL) and acetic acid
(150 mL) was dissolved in a mixed solution,
2,2,6,6-tetramethylpiperidine -N- oxyl at 30 ° C. It was added (966mg)
and ortho-periodic acid (16.9g), and the mixture was stirred for 1 hour
at 30-35 ℃. The reaction mixture was added 10% aqueous sodium bisulfite
solution (150 mL), after stirring for 30 minutes at room temperature,
toluene was added (400 mL), and concentrated in vacuo to 300 mL. The
solution further by the addition of toluene (400 mL), after
concentration under reduced pressure again to 300 mL, was added toluene
(500 mL), water (200 mL) and 50% aqueous sodium hydroxide (118 mL). Were
separated, the organic layer was washed with 20% brine (150 mL),
addition of toluene (200 mL), under reduced pressure and dehydrated
concentrated prepared in toluene (400 mL) solution. The compound
obtained in the solution (1-1) (116.5g), N, N- dimethylformamide (175
mL) and acetic acid (4.2 mL) was added, under reduced pressure, and
dried for 8 hours under reflux. The reaction was cooled to room
temperature, adding toluene (400 mL), washed once with 3 times with 5%
aqueous sodium bicarbonate solution (400 mL) and 10% brine (250 mL),
under reduced pressure and the organic layer was dried concentrated
toluene It was prepared (900 mL) solution. This solution was added
activated charcoal (15 g) at 35 ~ 40 ° C., after stirring for 30 minutes
at the same temperature, filtered and the activated carbon was washed
with toluene. Meet the filtrate and washings, after which was
concentrated under reduced pressure until 250mL, it was added dropwise
heptane (500mL) at room temperature. After stirring for 1.5 hours at the
same temperature, then cooled to 0 ℃, and the mixture was stirred for 1
hour. The precipitated crystals were filtered to give after washing
with toluene / heptane (1/2) mixture solution, the title compound was
dried under reduced pressure (85.0 g, 81.5% yield).
1 H-NMR (CDCl 3
) [delta]: 7.59 (1H, s), 7.47 (1H, s), 7.15 (1H, s), 7.08 (1H, brs),
3.92- 3.87 (1H, m), 3.78 (3H, s), 3.16-3.12 (2H, m), 2.96 (2H, t, J =
7.5Hz), 2.14- 2.11 (2H, m), 1.90-1.87 (2H, m), 1.77-1.65 (5H, m), 1.47
(9H, s), 1.17-1. 10 (2H, m), 0.96 (3H, d, J = 6.5Hz).
(1-4)
(2S) -5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans-4-
methylcyclohexyl)-lH-imidazol-4-yl] methyl} valerate (S )
-2-amino-1-propanol salt (A1 process, A2 process, A3 process)
[Of 14]

The
compound obtained in (1-3) (40.0g), (R) -2,2'- bis (di-3,5-xylyl)
-1,1'-binaphthyl (507.4Mg) and dichloro (p- cymene) ruthenium (II)
(dimer) and (211.4mg), were dissolved in degassed 2,2,2 trifluoroethanol
(400 mL), hydrogen under pressure (400-450kPa) , and the mixture was
stirred for 24 hours at 60 ℃. The reaction was cooled to room
temperature, after nitrogen substitution, and then concentrated under
reduced pressure to 60 mL.Tetrahydrofuran (200 mL) was added, was
concentrated under reduced pressure to 120 mL, of tetrahydrofuran was
added (200 mL).
To
the resulting solution was added water (160mL), cooled to 0 ℃, was
added a 50% aqueous solution of sodium hydroxide (24.0mL). After
stirring the reaction mixture at room temperature for 26 hours, and the
addition of 50% sodium hydroxide solution (8.00mL), and the mixture was
stirred for a further 4 hours. The reaction mixture under ice-cooling
was added dropwise concentrated hydrochloric acid (28 mL), activated
carbon was added (2.0 g) was stirred at room temperature for 10 minutes.
The active carbon was filtered off, washed with tetrahydrofuran / water
(2/1) mixed solvent (180 mL), sodium chloride (40 g) was separated by
adding and re-extract the aqueous layer with tetrahydrofuran (400 mL).
The organic layer was matched, and concentrated in vacuo to 200 mL.
After addition of toluene (400 mL) to this solution, under reduced
pressure and dehydrated concentrated prepared in toluene (200 mL)
solution.
After
adding tetrahydrofuran (400 mL) to the resulting solution was added (S)
-2- amino-1-propanol (8.2 g) at room temperature and stirred for 3
hours. The solution was cooled to 0 ℃, and was filtered after stirring
for 1.5 hours, it was precipitated crystals. The crystals were washed
with tetrahydrofuran and dried under reduced pressure to give the title
compound (45.4g, 98.2% yield, optical purity: ee 97.5%) was obtained.
1 H-NMR (CD
3 OD) [delta]: 7.57 (1H, s), 6.94 (1H, s), 3.98-3.85 (1H, yd),
3.69-3.64 ( 1H, m), 3.47-3.42 (1H , m), 3.29-3.23 (1H, m), 3.01 (2H, t, J
= 6.5Hz), 2.84 ( 1H, dd, J = 14.6,8.4Hz) , 2.55 (1H, dd, J =
14.6,6.2Hz), 2.52-2.45 (1H, m), 2.03 (2H, d, J = 12.7Hz ), 1.83 (2H, d, J
= 13.3Hz), 1.71 (2H, q, J = 12.5Hz), 1.60-1.44 ( 5H, m), 1.41 (9H , s),
1.23-1.20 (3H, m), 1.18-1.09 (2H, m), 0.94 (3H, d, J = 6.8Hz).
(1-5)
(2S) -5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans-4-
methylcyclohexyl)-lH-imidazol-4-yl] methyl} valerate (A4 process)
[Of 15]

(1-4)
The compound obtained in (40.0 g) in tetrahydrofuran (400 mL) and
dissolved in a mixed solvent of water (160 mL), concentrated
hydrochloric acid (7.3 mL) and added separation of sodium chloride (40
g) and washed 3 times with the organic layer 20% (w / w) brine (160 mL).
The organic layer under reduced pressure, dehydrated concentrated
prepared in toluene (320 mL) solution was dissolved after addition of
tetrahydrofuran (80 mL) was warmed precipitated 83 ° C. crystal. After
stirring overnight and cooled to room temperature, and stirred for a
further 3 hours at 0 ℃, and filtered the precipitated crystals. After
washing the crystals with toluene / tetrahydrofuran (4/1) mixed
solution, and dried under reduced pressure to give the title compound
(30.9g, 92.1% yield, optical purity: 97.4% ee) was obtained.
1 H-NMR (CDCl 3
) [delta]: 7.59 (1H, s), 6.73 (1H, s), 4.67 (1H, brs), 3.85-3.80 (1H,
yd), 3.12-3.08 (2H, m), 2.88 (1H, dd, J = 15.2,8.8Hz), 2.79 (1H, dd, J =
15.2,3.6Hz) , 2.70-2.64 (1H, m), 2.13-2.06 (2H, m), 1.90-1.82 (2H, m),
1.79-1.52 (5H, m), 1.49-1.44 (2H, m ), 1.43 (9H, s), 1.15-1.05 (2H, m),
0.95 (3H, d, J = 6. 5Hz).
(1-6) (2S) -5- amino -2 - {[1- (trans-4- methylcyclohexyl)-lH-imidazol-4-yl] methyl} valerate p- toluenesulfonate (A5 Step)
[Of 16]

In
tetrahydrofuran (100 mL), was dissolved the compound obtained in (1-5)
(25.0 g) and p- toluenesulfonic acid monohydrate (13.3 g), activated
charcoal (1 to this solution. 25 g) was added and stirred for 1 hour at
20 ~ 30 ℃. The charcoal was filtered and washed with tetrahydrofuran (50
mL).It matches the filtrate and washings, p- toluenesulfonic acid
monohydrate (13.3 g) and water (7.5 mL) and the mixture was heated under
reflux for 6 hours. The reaction was cooled to room temperature, it was
added triethylamine (7.7 g), at room temperature and stirred overnight.
To the reaction solution was added dropwise tetrahydrofuran (350 mL),
after stirring for 3 hours at room temperature and filtered the
precipitated crystal. After washing with tetrahydrofuran / water (50/1)
mixed solution, and dried under reduced pressure to give the title
compound (27.7g, 93.5% yield, optical purity: 98.4% ee) was obtained.
1 H-NMR (CD
3 OD) [delta]: 8.18 (1H, s), 7.70 (2H, d-, J = 8.1 Hz), 7.22 (2H, d-, J
= 7.5 Hz), 7.16 (1H, s), 4.06 (1H, tt, J = 12.0,3.9Hz), 2.94-2.86 (3H,
m), 2.69 (1H, dd, J = 14.6,5.8Hz), 2.62-2.59 (1H, m), 2.36 (3H, s),
2.08-2.05 (2H, m), 1.86-1 .83 (2H, m), 1.76-1.46 (7H, m), 1.18-1.11 (2H,
m), 0.94 (3H, d, J = 6.5Hz).
(Example
2) (2-1) (2S) -5 - [(tert-butoxycarbonyl) amino] -2 - {[1- (trans -4- methylcyclohexyl)-lH-imidazol-4-yl] methyl } methyl valerate
2) (2-1) (2S) -5 - [(tert-butoxycarbonyl) amino] -2 - {[1- (trans -4- methylcyclohexyl)-lH-imidazol-4-yl] methyl } methyl valerate
[Of 17]

It
was asymmetrically reduced using a number of catalysts. The reaction
conversion and the optical purity of the obtained title compound was
determined by the following HPLC analysis conditions.
Reaction conversion rate measurement:
Column: Waters XBridge C18 4.6mmI. D. × 150mm (3.5μm),
mobile phase: (A) 10mM aqueous ammonium acetate solution, (B)
acetonitrile, Gradient conditions: B: conc. ; 20% (0-5 minutes), 20-90% (5-20 minutes), 90% (20-24 minutes),
temperature: 40 ℃,
flow rate: 1.0mL / min,
detection method: UV at 215nm
retention time: raw material: 21.1 minutes, the product: 19.1 minutes,
(peak area of peak area + product of raw materials) peak area / of the reaction conversion rate = product.
Column: Waters XBridge C18 4.6mmI. D. × 150mm (3.5μm),
mobile phase: (A) 10mM aqueous ammonium acetate solution, (B)
acetonitrile, Gradient conditions: B: conc. ; 20% (0-5 minutes), 20-90% (5-20 minutes), 90% (20-24 minutes),
temperature: 40 ℃,
flow rate: 1.0mL / min,
detection method: UV at 215nm
retention time: raw material: 21.1 minutes, the product: 19.1 minutes,
(peak area of peak area + product of raw materials) peak area / of the reaction conversion rate = product.
Optical purity measurement conditions:
column: CHIRALPAK IA 4.6mmI. D. × 250mm (5μm),
mobile phase: ethanol / hexane = 20/80
Temperature: 35 ℃,
flow rate: 1.0mL / min,
detection method: UV at 210nm,
retention time: R body: 6.8 minutes, S body: 7.8 minutes.
column: CHIRALPAK IA 4.6mmI. D. × 250mm (5μm),
mobile phase: ethanol / hexane = 20/80
Temperature: 35 ℃,
flow rate: 1.0mL / min,
detection method: UV at 210nm,
retention time: R body: 6.8 minutes, S body: 7.8 minutes.
Daiichi Sankyo Company,Limited, 第一三共株式会社
WO2011115064.....
http://www.google.co.in/patents/WO2011115064A1?cl=en
[Reference Example 1] 5 - [(tert- butoxycarbonyl) amino] -2- (diethoxyphosphoryl) valeric acid tert- butyl

1 H-NMR (CDCl 3) δ: 1.31-1.36 (6H, m), 1.44 (9H, m), 1.48 (9H, m), 1.51-1.59 (2H, m), 1.78-2.00 (2H, m) , 2.83 (1H, ddd, J = 22.9, 10.7, 4.4 Hz), 3.06-3.18 (2H, m), 4.10-4.18 (4H, m), 4.58 (1H, br).
[Reference Example 2] 5 - [(tert- butoxycarbonyl) amino] -2- (1H- imidazol-4-ylmethyl) valeric acid tert- butyl

1 H-NMR (CDCl 3) δ: 1.41 (9H, s), 1.44 (9H, s), 1.48-1.57 (3H, m), 1.57-1.66 (1H, m), 2.58-2.68 (1H, m) , 2.73 (1H, dd, J = 14.7, 5.3 Hz), 2.89 (1H, dd, J = 14.7, 8.4 Hz), 3.02-3.19 (2H, m), 4.67 (1H, br s), 6.79 (1H, s), 7.54 (1H, s).
[Reference Example 3] 5 - [(tert- butoxycarbonyl) amino] -2- (methoxycarbonyl) valeric acid

1 H-NMR (CDCl 3) δ: 1.44 (9H, m), 1.50-1.60 (2H, m), 1.86-2.01 (2H, m), 3.07-3.20 (2H, m), 3.43 (1H, m) , 3.77 (3H, s), 4.64 (1H, br).
[Reference Example 4] 1- (trans-4- methylcyclohexyl) -1H- imidazole-4-carbaldehyde [Step 1] 1- (trans-4- methylcyclohexyl) -1H- imidazole-4-carboxylic acid ethyl

1 H-NMR (CDCl 3) δ: 0.96 (3H, d, J = 6.6 Hz), 1.13 (2H, m), 1.39 (3H, d, J = 7.0 Hz), 1.47 (1H, m), 1.68 ( 2H, m), 1.88 (2H, m), 2.12 (2H, m), 3.91 (1H, tt, J = 12.1, 3.9 Hz), 4.36 (2H, q, J = 7.0 Hz), 7.54 (1H, s ), 7.66 (1H, s).
[Step 2] [1- (trans-4- methylcyclohexyl) -1H- imidazole-4-yl] methanol

1 H-NMR (CDCl 3) δ: 0.95 (3H, d, J = 6.6 Hz), 1.04-1.17 (2H, m), 1.44 (1H, m), 1.59-1.73 (2H, m), 1.81-1.89 (2H, m), 2.04-2.13 (2H, m), 2.78 (1H, br), 3.84 (1H, tt, J = 12.1, 3.9 Hz), 4.59 (2H, s), 6.91 (1H, s), 7.49 (1H, s).
[Step 3] 1- (trans-4- methylcyclohexyl) -1H- imidazole-4-carbaldehyde

1 H-NMR (CDCl 3) δ: 0.97 (3H, d, J = 6.8 Hz), 1.09-1.19 (2H, m), 1.48 (1H, m), 1.65-1.75 (2H, m), 1.87-1.93 (2H, m), 2.11-2.18 (2H, m), 3.95 (1H, tt, J = 12.2, 3.9 Hz), 7.62 (1H, s), 7.68 (1H, s), 9.87 (1H, s).
[Example 15] (2R) -5- amino -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazole-4-yl] methyl} valeric acid and (2S) -5- amino-2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazol-4-yl] methyl} valeric acid [Step 1] 5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans 4-methylcyclohexyl) -1H- imidazole-4-yl] methyl} methyl valerate

1 H-NMR (CDCl 3) δ: 0.94 (3H, d, J = 6.6 Hz), 1.02-1.15 (2H, m), 1.34-1.69 (7H, m), 1.43 (9H, s), 1.80-1.87 (2H, m), 1.99-2.09 (2H, m), 2.69 (1H, dd, J = 13.7, 6.3 Hz), 2.79 (1H, m), 2.88 (1H, dd, J = 13.7, 7.4 Hz), 3.03-3.13 (2H, m), 3.63 (3H, s), 3.79 (1H, tt, J = 12.1, 3.9 Hz), 4.76 (1H, br), 6.67 (1H, s), 7.47 (1H, s) .
[Step 2] (2R) -5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazol-4-yl] methyl} methyl valerate and ( 2S) -5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazol-4-yl] methyl} methyl valerate

The solvent of the divided solution was evaporated under reduced pressure to give both enantiomers each (15mg). Both enantiomers were confirmed to be optically pure by analytical HPLC. Column: CHIRALPAK IA (0.46cm × 25.0cm), flow rate: 1mL / min, elution solvent: hexane / ethanol = 80/20 <v / v>, detection wavelength: 220nm, retention time: (2R) -5- [(tert- butoxycarbonyl) amino] -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazol-4-yl] methyl} methyl valerate (7.2 min), (2S) -5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazol-4-yl] methyl} methyl valerate (11.2 min).
[Step 3] (2R) -5- amino -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazole-4-yl] methyl} valerate

[Step 4] (2S) -5- amino -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazole-4-yl] methyl} valerate

[Example 16] 5-amino -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazole-4-yl] methyl} valeric acid benzyl hydrochloride [Step 1] 5 - [(tert- butoxycarbonyl) amino] -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazol-4-yl] methyl} valerate

MS (ESI) m / z 394 [M + H] +.
[Example 40] (2S) -5- Amino -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazol-4-yl] methyl} valerate · p- toluenesulfonate, anhydrous

1 H-NMR (CD 3 OD) δ: 0.95 (3H, d, J = 6.5 Hz), 1.11-1.21 (2H, m), 1.43-1.79 (7H, m), 1.83-1.89 (2H, m), 2.05-2.10 (2H, m), 2.37 (3H, s), 2.57-2.64 (1H, m), 2.70 (1H, dd, J = 14.5, 5.5 Hz), 2.85-2.95 (3H, m), 4.07 ( 1H, tt, J = 11.7, 3.9 Hz), 7.18 (1H, s), 7.23 (2H, d, J = 7.8 Hz), 7.70 (2H, d, J = 8.2 Hz), 8.22 (1H, s).
Elemental analysis: C 16 H 27 N 3 O 2 · C 7 H 8 O 3 S,
Theoretical value: C; 59.33, H; 7.58, N; 9.02, O; 17.18, S; 6.89,
Measured value: C; 59.09, H; 7.53, N; 8.92, O; 17.22, S; 6.78.
----------------------------------.
[Example 41] (2S) -5- Amino -2 - {[1- (trans-4- methylcyclohexyl) -1H- imidazol-4-yl] methyl} valerate · p- toluenesulfonate & 1 Water hydrate

Elemental analysis: C 16 H 27 N 3 O 2 · C 7 H 8 O 3 S · 1H 2 O,
Theoretical value: C; 57.12, H; 7.71, N; 8.69, O; 19.85, S; 6.63,
Measured value: C; 56.90, H; 7.69, N; 8.67, O; 19.81, S; 6.42.
References
Study to Assess the Safety, Pharmacokinetics, and Pharmacodynamics of DS-1040b in Subjects With Acute Ischemic Stroke (NCT02586233
Phase I Study to Evaluate the Safety and Tolerability of DS-1040b Intravenous Infusion With Clopidogrel in Healthy Subjects (NCT02560688)
Study of the Effects of Ethnicity on the Pharmacokinetics, Pharmacodynamics and Safety of DS-1040b (NCT02647307)
Edo, N.; Noguchi, K.; Ito, Y.; Morishima, Y.; Yamaguchi, K.
Hemorrhagic risk assessment of DS-1040 in a cerebral ischemia/reperfusion model of rats with hypertension and hyperglycemia
41st Int Stroke Conf (February 17-19, Los Angeles) 2016, Abst TP283
Noguchi, K.; Edo, N.; Ito, Y.; Morishima, Y.; Yamaguchi, K.
Improvement of cerebral blood flow with DS-1040 in a rat thromboembolic stroke model
41st Int Stroke Conf (February 17-19, Los Angeles) 2016, Abst TP271
Lapchak, P.A.; Boitano, P.D.; Noguchi, K.
DS-1040 an inhibitor of the activated thrombin activatable fibrinolysis inhibitor improves behavior in embolized rabbits
41st Int Stroke Conf (February 17-19, Los Angeles) 2016, Abst WP282
A first-in-human, single ascending dose study of DS-1040, an inhibitor of the activated form of thrombinactivatable fibrinolysis inhibitor (TAFIa), in healthy subjects
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO621-MON
Dow, J.; Puri, A.; McPhillips, P.; Orihashi, Y.; Dishy, V.; Zhou, J.
A drug-drug interaction study of DS-1040 and aspirin in healthy subjects
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO603-TUE
Noguchi, K.; Edo, N.; Ito, Y.; Yamaguchi, K.
Effect of DS-1040 on endogenous fibrinolysis and impact on bleeding time in rats
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst AS145
Noguchi, K.; Edo, N.; Ito, Y.; Maejima, T.; Yamaguchi, K.
DS-1040: A novel selective inhibitor of activated form of thrombin-activatable fibrinolysis inhibitor
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO203-MON
DS1040b/Aspirin Drug/Drug Interaction Study (NCT02071004)
ClinicalTrials.gov Web Site 2014, February 26
| Patent ID | Date | Patent Title |
|---|---|---|
| US2014178349 | 2014-06-26 | Cycloalkyl-Substituted Imidazole Derivative |
| US8609710 | 2013-12-17 | Cycloalkyl-substituted imidazole derivative |
//////DS-1040, DS 1040, phase 2, Daiichi Sankyo Co Ltd, Ischemic stroke
O=C(O)[C@@H](CCCN)Cc1cn(cn1)[C@@H]2CC[C@@H](C)CC2
O=S(=O)(O)c1ccc(C)cc1.O=C(O)[C@@H](CCCN)Cc1cn(cn1)[C@@H]2CC[C@@H](C)CC2
No comments:
Post a Comment