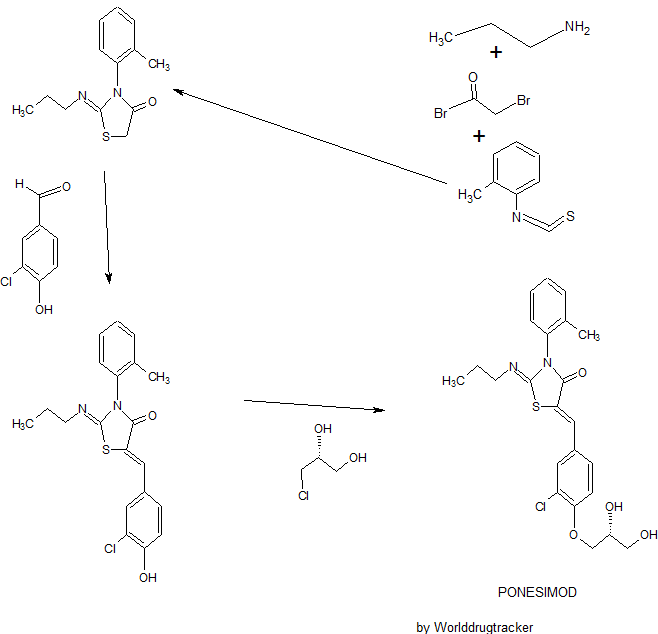
Ponesimod
Phase III
MW 460.97, C23 H25 Cl N2 O4 S
A sphingosine-1-phosphate receptor 1 (S1P1) agonist potentially for the treatment of multiple sclerosis.
(Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one
- (2Z,5Z)-5-[[3-Chloro-4-[(2R)-2,3-dihydroxypropoxy]phenyl]methylene]-3-(2-methylphenyl)-2-(propylimino)-4-thiazolidinone
- 5-[3-Chloro-4-[((2R)-2,3-dihydroxypropyl)oxy]benz-(Z)-ylidene]-2-((Z)-propylimino)-3-(o-tolyl)thiazolidin-4-one
- ACT 128800
ACT-128800; RG-3477; R-3477
CAS No. 854107-55-4
SYNTHESIS
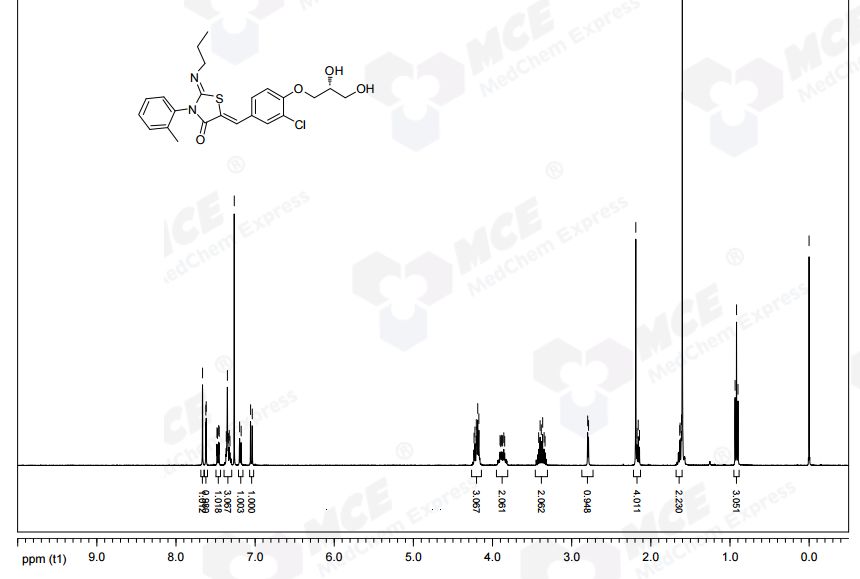
NMR CDCL3 FROM NET
Ponesimod (INN, codenamed ACT-128800) is an experimental drug for the treatment of multiple sclerosis (MS) and psoriasis. It is being developed by Actelion.
The first oral treatment for relapsing multiple sclerosis, the nonselective sphingosine-1-phosphate receptor (S1PR) modulator fingolimod, led to identification of a pivotal role of sphingosine-1-phosphate and one of its five known receptors, S1P1R, in regulation of lymphocyte trafficking in multiple sclerosis. Modulation of S1P3R, initially thought to cause some of fingolimod’s side effects, prompted the search for novel compounds with high selectivity for S1P1R. Ponesimod is an orally active, selective S1P1R modulator that causes dose-dependent sequestration of lymphocytes in lymphoid organs. In contrast to the long half-life/slow elimination of fingolimod, ponesimod is eliminated within 1 week of discontinuation and its pharmacological effects are rapidly reversible. Clinical data in multiple sclerosis have shown a dose-dependent therapeutic effect of ponesimod and defined 20 mg as a daily dose with desired efficacy, and acceptable safety and tolerability. Phase II clinical data have also shown therapeutic efficacy of ponesimod in psoriasis. These findings have increased our understanding of psoriasis pathogenesis and suggest clinical utility of S1P1R modulation for treatment of various immune-mediated disorders. A gradual dose titration regimen was found to minimize the cardiac effects associated with initiation of ponesimod treatment. Selectivity for S1P1R, rapid onset and reversibility of pharmacological effects, and an optimized titration regimen differentiate ponesimod from fingolimod, and may lead to better safety and tolerability. Ponesimod is currently in phase III clinical development to assess efficacy and safety in relapsing multiple sclerosis. A phase II study is also ongoing to investigate the potential utility of ponesimod in chronic graft versus host disease.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Biology and pharmacology of sphingosine-1-phosphate receptor 1
The past decades have witnessed major advances in the treatment of autoimmune and chronic inflammatory diseases. A plethora of novel therapies targeting specific molecules involved in the inflammatory or immune system activation cascades have become available. These have significantly increased our understanding of disease pathogenesis and improved the management of immune-mediated disorders. However, most of the targeted therapies are biological drugs which need to be injected, are eliminated slowly (e.g. over several weeks) and can lose efficacy or tolerability due to their potential immunogenicity. In an attempt to overcome these hurdles, pharmaceutical research has made considerable efforts to develop novel oral targeted therapies for autoimmune and chronic inflammatory diseases.
Sphingosine-1-phosphate receptor 1 (S1P1R) is one of five known G protein-coupled receptors with nanomolar affinity for the lysophospholipid sphingosine-1-phosphate (S1P), which is generated through physiologic metabolism of the cell membrane constituent sphingomyelin by all cells [Brinkmann, 2007]. S1P receptors, including S1P1R, are widely expressed in many tissues [Chun et al. 2010]. S1P1R expression on lymphocytes controls their egress from thymus and secondary lymphoid organs [Cyster and Schwab, 2012]. Lymphocyte egress requires a gradient of S1P concentration, which is established by a high S1P concentration in blood and lymph compared with a low concentration in the interstitial fluid of lymphoid organs [Grigorova et al. 2009].
Synthetic S1P1 receptor modulators disrupt the interaction of the physiologic S1P ligand with S1P1R by promoting initial activation followed by sustained internalization and desensitization of S1P1R [Hla and Brinkmann, 2011; Pinschewer et al. 2011]. Experiments conducted in animal models of transplant rejection, multiple sclerosis, lupus erythematosus, arthritis and inflammatory bowel disease with the first-generation, nonselective S1P receptor modulator, fingolimod, have demonstrated the potential efficacy of this mode of action across several immune-mediated chronic inflammatory conditions [Brinkmann, 2007]. Fingolimod is a structural analog of sphingosine that is phosphorylated in the body by a sphingosine kinase to generate the bioactive form of the drug, fingolimod phosphate, which binds to multiple S1P receptors [Brinkmann, 2007]. Clinical trials in multiple sclerosis (MS) have confirmed the efficacy of fingolimod in relapsing MS, but not in primary progressive disease, and led to the approval of the first oral medication for the treatment of relapsing forms of MS in 2010 [Kappos et al. 2010].
The mechanism of action of fingolimod has increased our understanding of MS pathogenesis. T and B cells, but not natural killer (NK) cells, express functional S1P1R and are affected by fingolimod [Cyster and Schwab, 2012]. Furthermore, S1P1R is differentially expressed and regulated in functionally distinct subsets of lymphocytes and fingolimod has been shown to predominantly affect naïve T cells and central memory T cells (TCM) while sparing effector memory T cells (TEM), and terminally differentiated effector T cells (TE) in patients with relapsing MS [Mehling et al. 2008, 2011]. This has raised the possibility that, at least in MS, retention of TCM cells, which include pro-inflammatory T helper 17 (Th17) cells, by fingolimod may prevent their accumulation in the cerebrospinal fluid (CSF) and subsequent differentiation to TE cells in the central nervous system (CNS) [Hla and Brinkmann, 2011]. The effects of S1P1R modulation on B cells are less well defined. Recent data from patients with relapsing MS have shown predominant reduction of memory B cells and recently activated memory B cells (CD38int-high) in peripheral blood after treatment with fingolimod [Claes et al. 2014; Nakamura et al. 2014]. As memory B cells are implicated in the pathogenesis of MS and other autoimmune diseases, these observations suggest another potential mechanism underlying the therapeutic effects of S1P1R modulators.
Astrocytes, microglia, oligodendrocytes and neurons express various S1P receptors including S1P1R, S1P3R and S1P5R. Fingolimod has been shown to penetrate the CNS tissues and in vitro studies have shown activation of astrocytes and oligodendrocytes by fingolimod [Foster et al. 2007]. Conditional deletion of S1P1R on neural cells in mice reduced the severity of experimental autoimmune encephalomyelitis (EAE) and reductions in the clinical scores were paralleled by decreased demyelination, axonal loss and astrogliosis [Choi et al. 2011]. Unfortunately, there was no beneficial effect in a recently completed, large study of fingolimod in patients with primary progressive MS [Lublin et al. 2015], suggesting that the direct effect on CNS cells alone may not be sufficient. Taken together, these data suggest the possibility of a direct beneficial effect of S1P1R modulation in the brain of patients with relapsing MS [Dev et al. 2008]; however, its contribution to efficacy relative to the immunological effects remains unclear.
Initial studies in rodents suggested that modulation of S1P3R on cardiac myocytes by fingolimod was associated with a reduction of heart rate (HR) by activation of G-protein-coupled inwardly rectifying potassium channels (GIRK) that regulate pacemaker frequency, and the shape and duration of action potentials [Koyrakh et al. 2005; Camm et al. 2014]. Modulation of S1P2R and S1P3R on myofibroblasts by fingolimod was also shown to stimulate extracellular matrix synthesis [Sobel et al. 2013]. Modulation of these receptors on vascular smooth muscle cells appeared to be associated with vasoconstriction, leading to the slight increase in blood pressure observed with fingolimod treatment [Salomone et al. 2003; Watterson et al. 2005; Hu et al. 2006; Lorenz et al. 2007; Kappos et al. 2010]. These observations raised the possibility that some side effects associated with fingolimod treatment could be avoided by more selective S1P1R modulators, thus triggering the search for novel compounds.
Currently, there are several selective S1P1R modulators in clinical development [Gonzalez-Cabrera et al.2014; Subei and Cohen, 2015]. Here we review data and the development status of ponesimod, a selective S1P1R modulator developed by Actelion Pharmaceuticals Ltd.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Ponesimod, a selective, rapidly reversible, orally active, sphingosine-1-phosphate receptor modulator
Ponesimod (ACT-128800 (Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one) is a selective, rapidly reversible, orally active, S1P1R modulator. Ponesimod emerged from the discovery of a novel class of S1P1R agonists based on the 2-imino-thiazolidin-4-one scaffold (Figure 1) [Bolli et al. 2010]. Ponesimod activates S1P1R with high potency [half maximal effective concentration (EC50) of 5.7 nM] and selectivity. Relative to the potency of S1P, the potency of ponesimod is 4.4 higher for S1P1R and 150-fold lower for S1P3R, resulting in an approximately 650-fold higher S1P1R selectivity compared with the natural ligand.

Chemical structure of ponesimod, C23H25N2O4CIS (molecular weight 460.98).http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Clinical trials
In a 2009–2011 Phase II clinical trial including 464 MS patients, ponesimod treatment resulted in fewer new active brain lesions thanplacebo, measured during the course of 24 weeks.[3][4]
In a 2010–2012 Phase II clinical trial including 326 patients with psoriasis, 46 or 48% of patients (depending on dosage) had a reduction of at least 75% Psoriasis Area and Severity Index (PASI) score compared to placebo in 16 weeks.[3][5]
Adverse effects
Common adverse effects in studies were temporary bradycardia (slow heartbeat), usually at the beginning of the treatment,dyspnoea (breathing difficulties), and increased liver enzymes (without symptoms). No significant increase of infections was observed under ponesimod therapy.[3] QT prolongation is detectable but was considered to be too low to be of clinical importance in a study.[6]
Mechanism of action
Like fingolimod, which is already approved for the treatment of MS, ponesimod blocks the sphingosine-1-phosphate receptor. This mechanism prevents lymphocytes (a type of white blood cells) from leaving lymph nodes.[3] Ponesimod is selective for subtype 1 of this receptor, S1P1.[7]
PAPER
2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1Receptor Agonists
Drug Discovery Chemistry, Actelion Pharmaceuticals Ltd., Gewerbestrasse 16, CH-4123 Allschwil, Switzerland
J. Med. Chem., 2010, 53 (10), pp 4198–4211
DOI: 10.1021/jm100181s
Publication Date (Web): May 06, 2010
Copyright © 2010 American Chemical Society
*To whom correspondence should be addressed. Phone: + 41 61 565 65 70. Fax: + 41 61 565 65 00. E-mail:martin.bolli@actelion.com.

Sphingosine-1-phosphate (S1P) is a widespread lysophospholipid which displays a wealth of biological effects. Extracellular S1P conveys its activity through five specific G-protein coupled receptors numbered S1P1 through S1P5. Agonists of the S1P1 receptor block the egress of T-lymphocytes from thymus and lymphoid organs and hold promise for the oral treatment of autoimmune disorders. Here, we report on the discovery and detailed structure−activity relationships of a novel class of S1P1 receptor agonists based on the 2-imino-thiazolidin-4-one scaffold. Compound 8bo (ACT-128800) emerged from this series and is a potent, selective, and orally active S1P1 receptor agonist selected for clinical development. In the rat, maximal reduction of circulating lymphocytes was reached at a dose of 3 mg/kg. The duration of lymphocyte sequestration was dose dependent. At a dose of 100 mg/kg, the effect on lymphocyte counts was fully reversible within less than 36 h. Pharmacokinetic investigation of8bo in beagle dogs suggests that the compound is suitable for once daily dosing in humans.
(Z,Z)-5-[3-Chloro-4-((2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolyl-thiazolidin-4-one (8bo)
..............DELETED............... column chromatography on silica gel eluting with heptane:ethyl acetate 1:4 to give the title compound (1.34 g, 37%) as a pale-yellow foam.
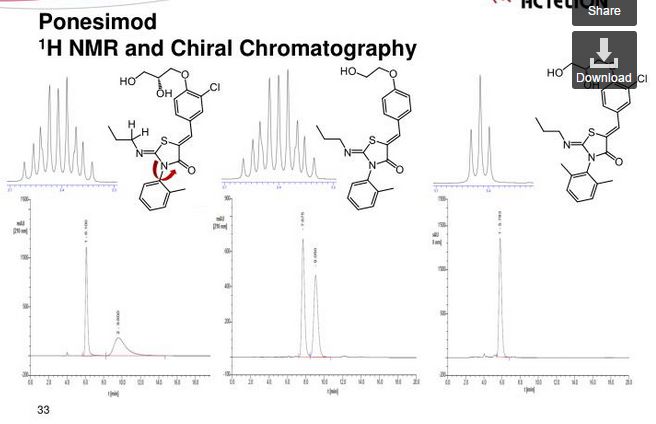
1H NMR (CDCl3): δ 0.94 (t, J = 7.3 Hz, 3 H), 1.58−1.70 (m, 2 H), 2.21 (s, 3 H), 3.32−3.48 (m, 2 H), 3.82−3.95 (m, 3 H), 4.12−4.27 (m, 4 H), 7.07 (d, J = 8.8 Hz, 1 H), 7.21 (d, J = 7.0 Hz, 1 H), 7.31−7.39 (m, 3 H), 7.49 (dd, J = 8.5, 2.0 Hz, 1 H), 7.64 (d, J= 2.0 Hz, 1 H), 7.69 (s, 1 H).
13C NMR (CDCl3): δ 11.83, 17.68, 23.74, 55.42, 63.46, 69.85, 70.78, 133.48, 120.75, 123.71, 127.05, 128.25, 128.60, 129.43, 130.06, 131.13, 131.50, 134.42, 136.19, 146.98, 154.75, 166.12. LC-MS (ES+): tR 0.96 min. m/z: 461 (M + H).
HPLC (ChiralPak AD-H, 4.6 mm × 250 mm, 0.8 mL/min, 70% hexane in ethanol): tR 11.8 min. Anal. (C23H25N2O4SCl): C, H, N, O, S, Cl.
PATENT
WO 2014027330
The present invention relates inter alia to a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (hereinafter also referred to as the "COMPOUND" or "compound (2)"), especially in crystalline form C which form is described in WO 2010/046835. The preparation of COMPOUND and its activity as immunosuppressive agent is described in WO 2005/054215. Furthermore, WO 2008/062376 describes a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one which can be used as an intermediate in the preparation of COMPOUND.
Example 1 a) below describes such a process of preparing (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one according to WO 2008/062376. According to WO 2008/062376 the obtained (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one can then be transformed into COMPOUND by using standard methods for the alkylation of phenols. Such an alkylation is described in Example 1 b) below. Unfortunately, this process leads to the impurity (2Z,5Z)-5-(3-chloro-4-((1 ,3-dihydroxypropan-2-yl)oxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one which is present in about 2% w/w in the crude product (see Table 1 ) and up to 6 recrystallisations are necessary in order to get this impurity below 0.4% w/w (see Tables 1 and 2) which is the specified limit based on its toxicological qualification.


the obtained (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one to form (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (2):

.
The reaction of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one can be performed under conditions which are typical for a Knoevenagel condensation. Such conditions are described in the literature for example in Jones, G., Knoevenagel Condensation in Organic Reaction, Wiley: New York, 1967, Vol. 15, p 204; or Prout, F. S., Abdel-Latif, A. A., Kamal, M. R., J. Chem. Eng. Data, 2012, 57, 1881-1886.
2-[(Z)-Propylimino]-3-o-tolyl-thiazolidin-4-one can be prepared as described in WO 2008/062376, preferably without the isolation and/or purification of intermediates such as the thiourea intermediate that occurs after reacting o-tolyl-iso-thiocyanate with n-propylamine. Preferably 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one obtained according to WO 2008/062376 is also not isolated and/or purified before performing the Knoevenagel condensation, i.e. before reacting 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one with (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ), i.e. in a preferred embodiment compound (2) is prepared in a one-pot procedure analogous to that described in WO 2008/062376.
Example 1 : (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one

a) Preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one:

Acetic acid solution: To acetic acid (149.2 mL) are added sodium acetate (1 1 .1 1 g, 2.00 eq.) and 3-chloro-4-hydroxybenzaldehyde (10.60 g, 1.00 eq.) at 20 °C. The mixture is stirred at 20 °C until complete dissolution (2 to 3 h).
n-Propylamine (4.04 g, 1.00 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 40 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (13.53 g, 1.00 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). 70 mL of dichloromethane are distilled off under atmospheric pressure and jacket temperature of 60 °C. The temperature is adjusted to 42 °C and the acetic acid solution is added to the reaction mixture. The resulting solution is heated to 58 °C and stirred at this temperature for 15 h before IPC (conversion specification≥ 95 %). 25 mL of solvents are distilled off under vacuum 900 - 500 mbars and jacket temperature of 80 °C. The temperature is adjusted to 60 °C and water (80.1 mL) is added to the reaction mixture over 1 h. The resulting yellow suspension is stirred at 60 °C for 30 min. The suspension is cooled to 20 °C over 1 h and stirred at this temperature for 30 min.
The product is filtered and washed with a mixture of acetic acid (30 mL) and water (16 mL) and with water (50 mL) at 20 °C. The product is dried under vacuum at 50 °C for 40 h to afford a pale yellow solid; yield 25.93 g (78 %).
b) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
To a suspension of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one (10.00 g, 1.00 eq.) in ethanol (47.2 mL) is added (R)-3-chloro-1 ,2-
propanediol (3.37 g, 1.18 eq.) at 20 °C. Potassium tert-butoxide (3.39 g, 1.13 eq.) is added in portions at 20 °C. The resulting fine suspension is stirred at 20 °C for 25 min before being heated to reflux (88 °C). The reaction mixture is stirred at this temperature for 24 h before IPC (conversion specification≥ 96.0 %). After cooling down to 60 °C, acetonitrile (28.6 mL) and water (74.9 mL) are added. The resulting clear solution is cooled from 60 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.010 g, 0.001 eq.; crystalline form C can be prepared as described in WO 2010/046835) are added at 50 °C. The suspension is heated from 0 °C to 50 °C, cooled to 0 °C over 6 h and stirred at this temperature for 12 h.
The product is filtered and washed with a mixture of acetonitrile (23.4 mL) and water (23.4 mL) at 0 °C. The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield 1 1.91 g (84 %).
c) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
Recrystallisation I: The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in acetonitrile (30 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 12.8 mL).
Recrystallisation II: The wet product is dissolved in acetonitrile (27.0 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 1 1.3 mL).
Recrystallisation III: The wet product is dissolved in acetonitrile (24.3 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4- one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 10.1 mL).
Recrystallisation IV: The wet product is dissolved in acetonitrile (21.9 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 9.1 mL).
Recrystallisation V: The wet product is dissolved in acetonitrile (19.7 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 8.2 mL).
Recrystallisation VI: The wet product is dissolved in acetonitrile (23.9 mL) at 70 °C. Water (20 mL) is added at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h.
During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2- (propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed twice with a mixture of acetonitrile (4.5 mL) and water (4.5 mL) at -10 °C.
The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield: 7.0 g (70 %).
Example 2: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde

Potassium tert-butoxide (1 18 g, 1.20 eq.) is added to n-propanol (963 mL) followed by 3-chloro-4-hydroxybenzaldehyde (137 g, 1.00 eq.). To the mixture is added (R)-3-chloro-1 ,2-propanediol (126 g, 1.30 eq.). The suspension is heated to 90 °C and stirred at this temperature for 17 h. Solvent (500 mL) is distilled off at 120 °C external temperature and reduced pressure. Water is added (1.1 L) and solvent (500 mL) is removed by distillation. The turbid solution is cooled to 20 °C. After stirring for one hour a white suspension is obtained. Water (500 mL) is added and the suspension is cooled to 10 °C. The suspension is filtered and the resulting filter cake is washed with water (500 mL). The product is dried at 50 °C and reduced pressure to yield 149 g of a white solid (73%), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 3: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde
Potassium tert-butoxide (8.60 g, 1.20 eq.) is added to n-propanol (70 mL) below 15 °C, the temperature is allowed to rise. After the addition the temperature is corrected again to below 15 °C before addition of 3-chloro-4-hydroxybenzaldehyde (10 g, 1 .00 eq.). The suspension is heated to 40 °C and stirred for 30 min. (R)-3-Chloro-1 ,2-propanediol (9.18 g, 1.30 eq.) is added at 40 °C. The resulting suspension is heated to 60 °C and stirred at this temperature for 15 h then heated to 94 °C till meeting the IPC-specification (specification conversion≥ 90.0 %). The mixture is cooled to 30 °C and n-propanol is partially distilled off (-50 mL are distilled off) under reduced pressure and a maximum temperature of 50 °C, the jacket temperature is not allowed to raise above 60 °C.
Water (81 mL) is added and a second distillation is performed under the same conditions (24 mL are distilled off). The mixture is heated till homogeneous (maximum 54 °C) and then cooled to 24 °C. At 24 °C the mixture is seeded with crystalline (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of form A (0.013 g, 0.00085 eq.). How to obtain the crystalline seeds is described in Examples 2 and 5. The reaction mixture is cooled to 0 °C over 7.5 h.
The product is filtered and washed with water (2 x 35 mL) and once with methyl tert-butyl ether (20 mL) at 5 °C. The product is dried under vacuum at 40 °C for 20 h to afford an off-white solid; yield: 10.6 g (72 %), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 4: (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)- 3-(o-tolyl)thiazolidin-4-one
a) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
n-Propylamine (5.23 g, 1.32 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 15 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (14.88 g, 1.10 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). Dichloromethane is partially distilled off (66 mL are distilled off) under atmospheric pressure and jacket temperature of 60 °C. Ethanol (1 1 1.4 mL), sodium acetate (12.75 g, 2.30 eq.) and (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde from Example 3 (14.38 g, 0.93 eq.) are added. The remaining dichloromethane and a part of ethanol are distilled off (49.50 mL are distilled off) under atmospheric pressure and jacket temperature up to 85 °C. The reaction mixture (orange suspension) is stirred for 3 - 5 h under reflux (78 °C) before IPC (conversion specification≥ 97.0 %).
Water (88.83 mL) is added and the temperature adjusted to 40 °C before seeding with micronized (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.075 g, 0.0024 eq.). The reaction mixture is cooled to 0 °C over 5 h, heated up to 40 °C, cooled to 0 °C over 6 h and stirred at this temperature for 2 h.
The product is filtered and washed with a 1 :1 ethanohwater mixture (2 x 48 mL) at 0 °C. The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 24.71 g (86 %).
b) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in ethanol (40 mL) at 70 °C. The temperature is adjusted at 50 °C for seeding with micronised (2Z,5Z)-5-(3-chloro-4-((R)-2,3- dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.016 g, 0.0016 eq.). The reaction mixture is cooled from 50 °C to 0 °C over 4 h, heated up to 50 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h.
The product is filtered and washed with ethanol at 0 °C (2 x 12.8 mL). The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 9.2 g (92 %).
Example 5: Preparation of crystalline seeds of (R)-3-chloro-4-(2,3-dihydroxypropoxy)- benzaldehyde
10 mg of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of at least 99.5% purity by 1 H-NMR assay is dissolved in a 4 mL vial by adding 1 mL of pure ethanol (puriss p. a.). The solvent is allowed to evaporate through a small hole in the cap (approx. 2 mm of diameter) of the vial until complete dryness. The white solid residue is crystalline (R)-3-chloro-4-(2,3- dihydroxypropoxy)-benzaldehyde in crystalline form A. Alternatively, methanol or methylisobutylketone (both in puriss p. a. quality) is used. This procedure is repeated until sufficient seeds are made available.

PATENT
WO 2005054215
| WO2005054215A1 | Nov 16, 2004 | Jun 16, 2005 | Actelion Pharmaceuticals Ltd | 5-(benz- (z) -ylidene) -thiazolidin-4-one derivatives as immunosuppressant agents |
| WO2008062376A2 | Nov 22, 2007 | May 29, 2008 | Actelion Pharmaceuticals Ltd | New process for the preparation of 2-imino-thiazolidin-4-one derivatives |
| WO2010046835A1 | Oct 19, 2009 | Apr 29, 2010 | Actelion Pharmaceuticals Ltd | Crystalline forms of (r) -5- [3-chloro-4- ( 2, 3-dihydroxy-propoxy) -benz [z] ylidene] -2- ( [z] -propylimino) -3-0-tolyl-thiazolidin-4-one |
| Reference | ||
|---|---|---|
| 1 | * | BOLLI, M.H. ET AL.: "2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1 Receptor Agonists", JOURNAL OF MEDICINAL CHEMISTRY, vol. 53, no. 10, 2010, pages 4198-4211, XP55090073, ISSN: 0022-2623, DOI: 10.1021/jm100181s |
References
- "Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: Favorable impact of dose up-titration". The Journal of Clinical Pharmacology 54: 179–88. Feb 2014. doi:10.1002/jcph.244. PMID 24408162.
- "Mass balance, pharmacokinetics and metabolism of the selective S1P1 receptor modulator ponesimod in humans". Xenobiotica 45: 139–49. Feb 2015. doi:10.3109/00498254.2014.955832. PMID 25188442.
- H. Spreitzer (29 September 2014). "Neue Wirkstoffe – Ponesimod". Österreichische Apothekerzeitung (in German) (20/2014): 42.
- "Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised phase II trial". Journal of Neurology, Neurosurgery 85: 1198–208. Nov 2014. doi:10.1136/jnnp-2013-307282. PMC 4215282. PMID 24659797.
- "Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double-blind, placebo-controlled phase 2 trial". The Lancet 384: 2036–45. Dec 2014. doi:10.1016/S0140-6736(14)60803-5. PMID 25127208.
- "Effect of Ponesimod, a selective S1P1 Receptor Modulator, on the QT Interval in Healthy Subjects". Basic 116: 429–37. May 2015.doi:10.1111/bcpt.12336. PMID 25287214.
- "Ponesimod". Actelion. Retrieved 31 October 2014.

ABOUT PONESIMOD
Ponesimod is a potent orally active, selective sphingosine-1-phosphate receptor 1 (S1P1) immunomodulator.
Ponesimod prevents lymphocytes from leaving lymph nodes, thereby reducing circulating blood lymphocyte counts and preventing infiltration of lymphocytes into target tissues. The lymphocyte count reduction is rapid, dose-dependent, sustained upon continued dosing, and quickly reversible upon discontinuation. Initial data suggest that ponesimod does not cause lymphotoxicity by destroying/depleting lymphocytes or interfering with their cellular function. Other blood cells e.g. cells of the innate immune system are largely unaffected. Ponesimod is therefore considered a promising new oral agent for the treatment of a variety of autoimmune disorders.
Ponesimod prevents lymphocytes from leaving lymph nodes, thereby reducing circulating blood lymphocyte counts and preventing infiltration of lymphocytes into target tissues. The lymphocyte count reduction is rapid, dose-dependent, sustained upon continued dosing, and quickly reversible upon discontinuation. Initial data suggest that ponesimod does not cause lymphotoxicity by destroying/depleting lymphocytes or interfering with their cellular function. Other blood cells e.g. cells of the innate immune system are largely unaffected. Ponesimod is therefore considered a promising new oral agent for the treatment of a variety of autoimmune disorders.
CURRENT STATUS
OPTIMUM (Oral Ponesimod versus Teriflunomide In relapsing MUltiple sclerosis) is a Phase III multi-center, randomized, double-blind, parallel-group, active-controlled superiority study to compare the efficacy and safety of ponesimod to teriflunomide in patients with relapsing multiple sclerosis (RMS). The study aims to determine whether ponesimod is more efficacious than teriflunomide in reducing relapses. The study is expected to enroll approximately 1’100 patients, randomized in 2 groups in a 1:1 ratio to receive ponesimod 20 mg/day or teriflunomide 14 mg/day, and is expected to last a little over 3 years. An additional study to further characterize the utility and differentiation of ponesimod in multiple sclerosis is being discussed with Health Authorities.
Ponesimod is also evaluated in a Phase II open-label, single-arm, intra-subject dose-escalation study to investigate the biological activity, safety, tolerability, and pharmacokinetics of ponesimod in patients suffering from moderate or severe chronic graft versus host disease (GvHD)inadequately responding to first- or second-line therapy. The study will also investigate the clinical response to ponesimod treatment in these patients. Approximately 30 patients will be enrolled to receive ponesimod in escalating doses of 5, 10, and 20 mg/day over the course of 24 weeks. The study is being conducted at approximately 10 sites in the US and is expected to last approximately 18 months.
Ponesimod is also evaluated in a Phase II open-label, single-arm, intra-subject dose-escalation study to investigate the biological activity, safety, tolerability, and pharmacokinetics of ponesimod in patients suffering from moderate or severe chronic graft versus host disease (GvHD)inadequately responding to first- or second-line therapy. The study will also investigate the clinical response to ponesimod treatment in these patients. Approximately 30 patients will be enrolled to receive ponesimod in escalating doses of 5, 10, and 20 mg/day over the course of 24 weeks. The study is being conducted at approximately 10 sites in the US and is expected to last approximately 18 months.
AVAILABLE CLINICAL DATA
The decision to move into Phase III development was based on the Phase IIb dose-finding study with ponesimod in patients with relapsing-remitting multiple sclerosis. A total of 464 patients were randomized into this study and the efficacy, safety and tolerability of three ponesimod doses (10, 20, and 40 mg/day) versus placebo, administered once daily for 24 weeks.
The primary endpoint of this study was defined as the cumulative number of new gadolinium-enhancing lesions on T1-weighted magnetic resonance imaging (MRI) scans at weeks 12, 16, 20, and 24 after study drug initiation. A key secondary endpoint of this study was the annualized relapse rate over 24 weeks of treatment. Patients who completed 24 weeks of treatment were offered the opportunity to enter into an extension study. This ongoing trial is investigating the long-term safety, tolerability, and efficacy of 10 and 20 mg/day of ponesimod in patients with relapsing-remitting multiple sclerosis, in a double-blind fashion. The study continues to provide extensive safety and efficacy information for ponesimod in this indication, with some patients treated for more than 6 years.
The safety database from all studies with ponesimod now comprises more than 1,300 patients and healthy volunteers.
The primary endpoint of this study was defined as the cumulative number of new gadolinium-enhancing lesions on T1-weighted magnetic resonance imaging (MRI) scans at weeks 12, 16, 20, and 24 after study drug initiation. A key secondary endpoint of this study was the annualized relapse rate over 24 weeks of treatment. Patients who completed 24 weeks of treatment were offered the opportunity to enter into an extension study. This ongoing trial is investigating the long-term safety, tolerability, and efficacy of 10 and 20 mg/day of ponesimod in patients with relapsing-remitting multiple sclerosis, in a double-blind fashion. The study continues to provide extensive safety and efficacy information for ponesimod in this indication, with some patients treated for more than 6 years.
The safety database from all studies with ponesimod now comprises more than 1,300 patients and healthy volunteers.
MILESTONES
2015 – Phase III program in multiple sclerosis initiated
2011 – Phase IIb dose-finding study in multiple sclerosis successfully completed
2006 – Entry-into-man
2004 – Preclinical development initiated
2011 – Phase IIb dose-finding study in multiple sclerosis successfully completed
2006 – Entry-into-man
2004 – Preclinical development initiated
KEY SCIENTIFIC LITERATURE
Olsson T et al. J Neurol Neurosurg Psychiatr. 2014 Nov;85(11):1198-208. doi: 10.1136/jnnp-2013-307282. Epub 2014 Mar 21
Freedman M.S, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 420 (P923).
Fernández Ó, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 417 (P919).
Piali L, Froidevaux S, Hess P, et al. J Pharmacol Exp Ther 337(2):547-56, 2011
Bolli MH, Abele S, Binkert C, et al. J Med Chem. 53(10):4198-211, 2010
Kappos L et al. N Engl J Med. 362(5):387-401, 2010
Freedman M.S, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 420 (P923).
Fernández Ó, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 417 (P919).
Piali L, Froidevaux S, Hess P, et al. J Pharmacol Exp Ther 337(2):547-56, 2011
Bolli MH, Abele S, Binkert C, et al. J Med Chem. 53(10):4198-211, 2010
Kappos L et al. N Engl J Med. 362(5):387-401, 2010
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
| (2Z,5Z)-5-{3-Chloro-4-[(2R)-2,3-dihydroxypropoxy]benzylidene}-3-(2-methylphenyl)-2-(propylimino)-1,3-thiazolidin-4-one | |
| Clinical data | |
| Routes of administration | Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | 2 main metabolites |
| Biological half-life | 31–34 hrs[1] |
| Excretion | Feces (57–80%, 26% unchanged), urine (10–18%)[2] |
| Identifiers | |
| CAS Number | 854107-55-4 |
| ATC code | none |
| PubChem | CID 11363176 |
| ChemSpider | 9538103 |
| ChEMBL | CHEMBL1096146 |
| Synonyms | ACT-128800 |
| Chemical data | |
| Formula | C23H25ClN2O4S |
| Molar mass | 460.974 g/mol |
////Ponesimod, Phase III , A sphingosine-1-phosphate receptor 1, S1P1 agonist, multiple sclerosis. ACT-128800; RG-3477; R-3477, autoimmune disease, lymphocyte migration, multiple sclerosis, psoriasis, transplantation
CCC/N=C\1/N(C(=O)/C(=C/C2=CC(=C(C=C2)OC[C@@H](CO)O)Cl)/S1)C3=CC=CC=C3C
- NMR
- Supporting Info

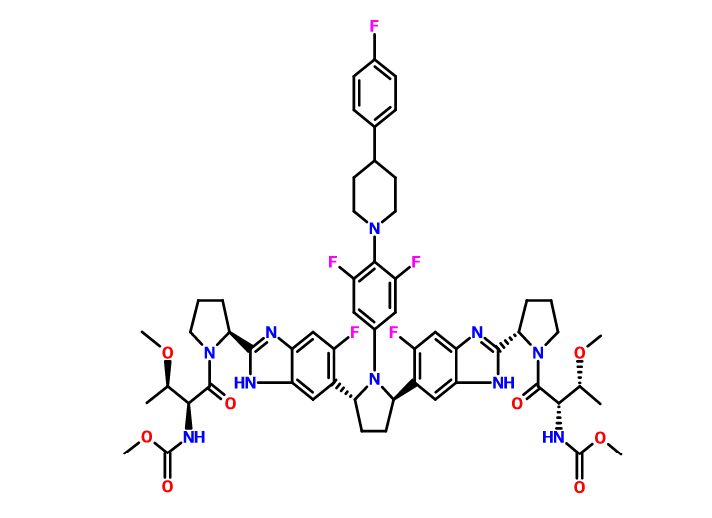

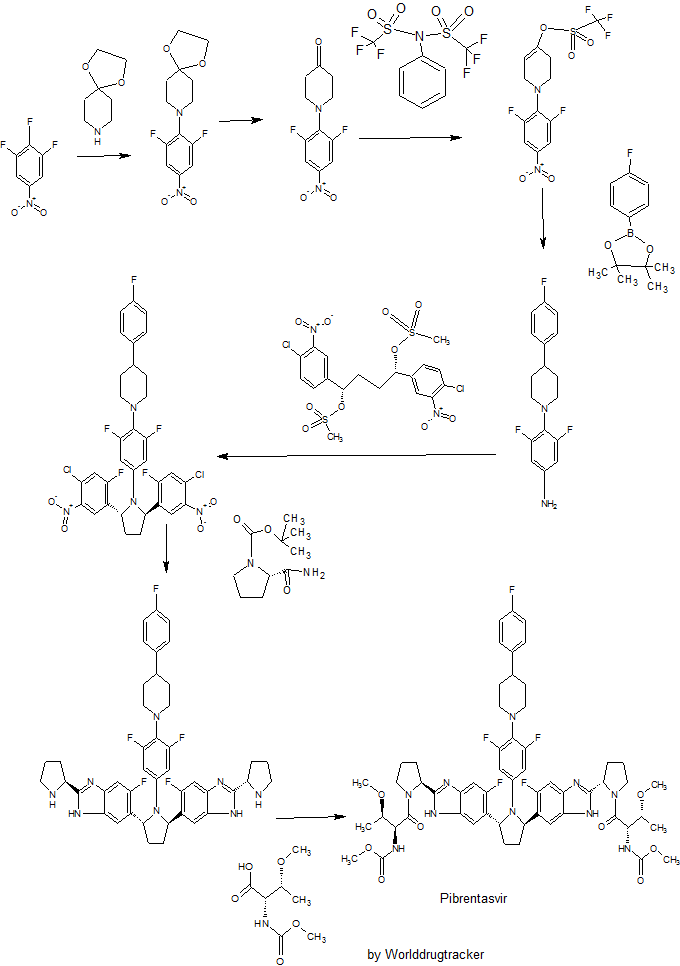
 Pibrentasvir
Pibrentasvir Pibrentasvir
Pibrentasvir