Rifaximin;
Rifaxidin; Rifacol; Xifaxan; Normix; Rifamycin L 105;L 105 (ansamacrolide antibiotic), L 105SV
(2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro[4,5-e]pyrido[1,2-á]-benzimidazole-1,15(2H)-dione,25-acetate
CAS 80621-81-4, 4-Deoxy-4-methylpyrido[1,2-1,2]imidazo[5,4-c]rifamycin SV,
4-Deoxy-4′-methylpyrido[1′,2′-1,2]imidazo[5,4-c]rifamycin SV, Rifacol
| C43H51N3O11 | |
| Molecular Weight: | 785.87854 g/mol |
|---|

XIFAXAN tablets for oral administration are film-coated and contain 200 mg or 550 mg of rifaximin.
Rifaximin is an orally administered, semi-synthetic, nonsystemic antibiotic derived from rifamycin SV with antibacterial activity. Rifaximin binds to the beta-subunit of bacterial DNA-dependent RNA polymerase, inhibiting bacterial RNA synthesis and bacterial cell growth. As rifaximin is not well absorbed, its antibacterial activity is largely localized to the gastrointestinal tract.

Rifaximin (trade names:RCIFAX, Rifagut, Xifaxan, Zaxine) is a semisynthetic antibiotic based on rifamycin. It has poor oral bioavailability, meaning that very little of the drug will be absorbed into the blood stream when it is taken orally. Rifaximin is used in the treatment of traveler's diarrhea, irritable bowel syndrome, and hepatic encephalopathy, for which it receivedorphan drug status from the U.S. Food and Drug Administration in 1998.
Rifaximin is a rifamycin that was launched in 1988 by Alfa Wasserman for the treatment of bacterial infection, and was commercialized in 2004 by Salix for the treatment of Clostridium difficile-associated diarrhea. In 2008, the product was launched in Germany for the treatment of travelers' diarrhea caused by non-invasive enteropathogenic bacteria in adults. In 2015, Xifaxan was approved in the U.S. for the treatment of abdominal pain and diarrhea in adult men and women with irritable bowel syndrome with diarrhea. At the same year, Aska filed an application for approval of the product in Japan for the treatment of hepatic encephalopathy.
Rifaximin is licensed by the U.S. Food and Drug Administration to treat traveler's diarrhea caused by E. coli.[1] Clinical trials have shown that rifaximin is highly effective at preventing and treating traveler's diarrhea among travelers to Mexico, with fewside effects and low risk of developing antibiotic resistance.[2][3][4] It is not effective against Campylobacter jejuni, and there is no evidence of efficacy against Shigella or Salmonella species.
| Launched - 1988 | Alfa Wassermann | Infection, bacterial |
| Launched - 2004 | Salix | Traveler's diarrhea |
| Launched - 2010 | Salix | Encephalopathy, hepatic |
| Launched - 2015 | Salix | Irritable bowel syndrome (Diarrhea predominant) |
| Launched | Alfa Wassermann Merck & Co. | Hyperammonemia |
The drug is also at Salix in phase II trials for the treatment of Crohn's disease. Alfa Wasserman is also conducting phase II trials for Crohn's disease. The product was approved and launched in the U.S. for the maintenance of remission of hepatic encephalopathy in 2010. Mayo Clinic is conducting phase II clinical trials in the U.S. for the treatment of primary sclerosing cholangitis and the University of Hong Kong is also conducting Phase II trials for the treatment of functional dyspepsia.
It may be efficacious in relieving chronic functional symptoms of bloating and flatulence that are common in irritable bowel syndrome (IBS),[5][6] especially IBS-D.
In February 1998, Salix was granted orphan drug designation by the FDA for the use of rifaximin to treat hepatic encephalopathy. In 2009, a codevelopment agreement was established between Lupin and Salix in the U.S. for the development of a new formulation using Lupin's bioadhesive drug delivery technology.
There was recentlya pilot-study done on the efficacy of rifaximin as a means of treatment for rosacea, according to the study, induced by the co-presence of small intestinal bacterial overgrowth.[7]
In the United States, rifaximin has orphan drug status for the treatment of hepatic encephalopathy.[8] Although high-quality evidence is still lacking, rifaximin appears to be as effective as or more effective than other available treatments for hepatic encephalopathy (such as lactulose), is better tolerated, and may work faster.[9] Hepatic encephalopathy is a debilitating condition for those with liver disease. Rifaximin is an oral medication taken twice daily that helps patients to avoid reoccurring hepatic encephalopathy. It has minimal side effects, prevents reoccurring encephalopathy and high patient satisfaction. Patients are more compliant and satisfied to take this medication than any other due to minimal side effects, prolong remission, and overall cost.[10] Rifaximin helps patients avoid multiple readmissions from hospitals along with less time missed from work as well. Rifaximin should be considered a standard prescribed medication for those whom have episodes of hepatic encephalopathy.
The drawbacks to rifaximin are increased cost and lack of robust clinical trials for HE without combination lactulose therapy.
Also treats hyperammonemia by eradicating ammoniagenic bacteria.
Mechanism of action
Rifaximin interferes with transcription by binding to the β-subunit of bacterial RNA polymerase.[11] This results in the blockage of the translocation step that normally follows the formation of the first phosphodiester bond, which occurs in the transcription process.[12]
Efficacy
A 2011 study in patients with IBS (sans constipation) indicated 11% showed benefits over a placebo.[13] The study was supported by Salix Pharmaceuticals, the patent holder.[13] A 2010 study in patients treated for Hepatic Cirrhosis with hospitalization involving Hepatic encephalopathy resulted in 22% of the rifaxmin treated group experiencing a breakthrough episode of Hepatic encephalopathy as compared to 46% of the placebo group. The majority patients were also receivingLactulose therapy for prevention of hepatic encephalopathy in addition to Rifaximin.[14] Rifaximin shows promising results, causing remission in up to 59% of people with Crohn’s disease and up to 76% of people with Ulcerative Colitis.[15]
Availability
In the United States, Salix Pharmaceuticals holds a US Patent for rifaximin and markets the drug under the name Xifaxan, available in tablets of 200 mg and 550 mg.[16][17] In addition to receiving FDA approval for traveler’s diarrhea and (marketing approved for)[17] hepatic encephalopathy, Xifaxan received FDA approval for IBS in May 2015.[18] No generic formulation is available in the US and none has appeared due to the fact that the FDA approval process was ongoing. If Xifaxan receives full FDA approval for hepatic encephalopathy it is likely that Salix will maintain marketing exclusivity and be protected from generic formulations until March 24, 2017.[17] Price quotes received on February 21, 2013 for Xifaxan 550 mg in the Denver Metro area were between $23.57 and $26.72 per tablet. A price quote received on June 24, 2016 for Xifaxan 550 mg was $31.37 per tablet.
Rifaximin is approved in 33 countries for GI disorders.[19][20] On August 13, 2013, Health Canada issued a Notice of Compliance to Salix Pharmaceuticals Inc. for the drug product Zaxine.[21] In India it is available under the brand names Ciboz and Xifapill.[
SPECTRA
LINK IS CLICK
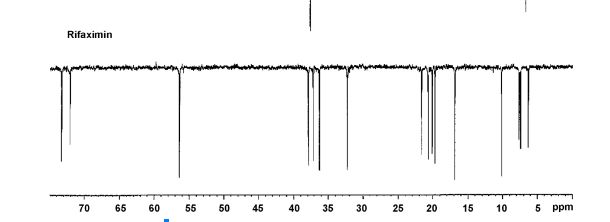
APT 13C NMR RIFAXIMIN
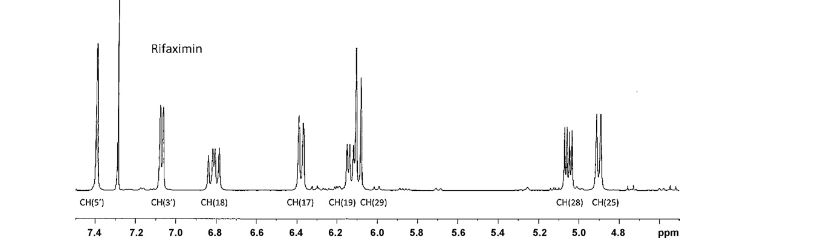
1H NMR PARTIAL
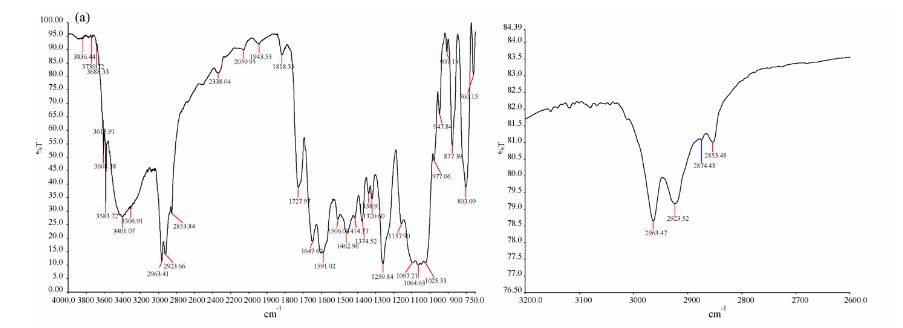
IR
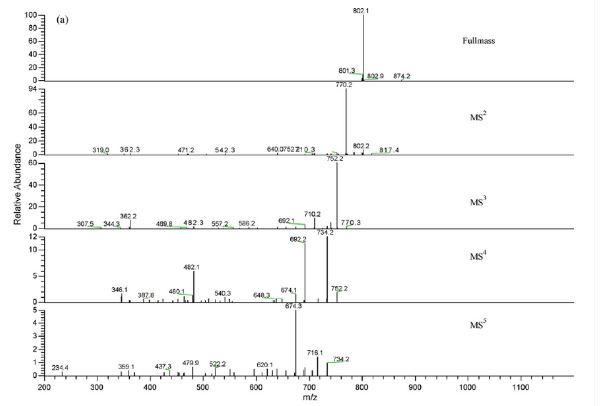
Direct infusion mass analysis ESI (+)
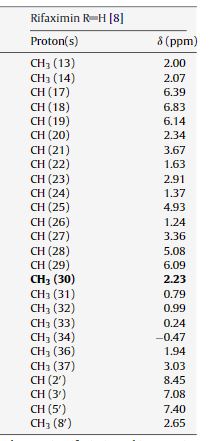
IH NMR
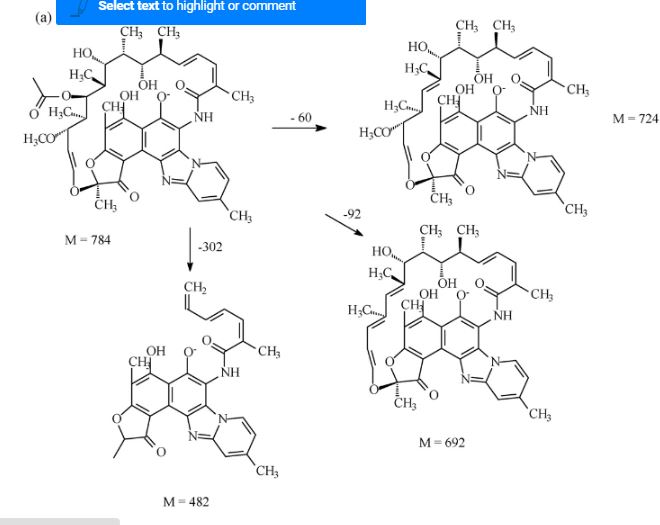
- [-]ESI FRAG PATHWAY
Synthesis
Rifaximin is a broad-spectrum antibiotic belonging to the family of Rifamycins and shows its antibacterial activity, in the gastrointestinal tract against localized bacteria that cause infectious diarrhoea, irritable bowel syndrome, small intestinal bacterial overgrowth, Crohn's disease, and/or pancreatic insufficiency.
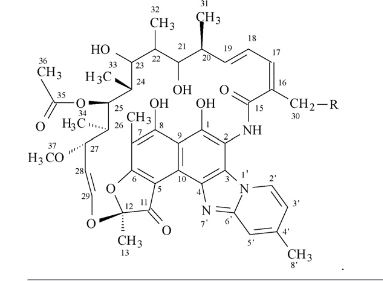
Rifaximin is sold under the brand name Xifaxan® in US for the treatment of Travellers' diarrhoea and Hepatic Encephalopathy. The chemical name of Rifaximin is (2S , 16Z, 18E,20S ,21 S ,22R,23R,24R,25S ,26S ,27S ,28E)-5,6,21 ,23 ,25-pentahydroxy-27-methoxy-2,4,1 l,16,20,22,24,26-octamethyl-2,7(epoxypentadeca-[l,l l,13]trienimino) benzofuro[4,5-e]pyrido[l,2-a]-benzimidazole-l,15(2H)-dione,25-acetate and the molecular formula is G^HsiNsOn with a molecular weight of 785.9. The structural formula of Rifaximin is:

Formula I
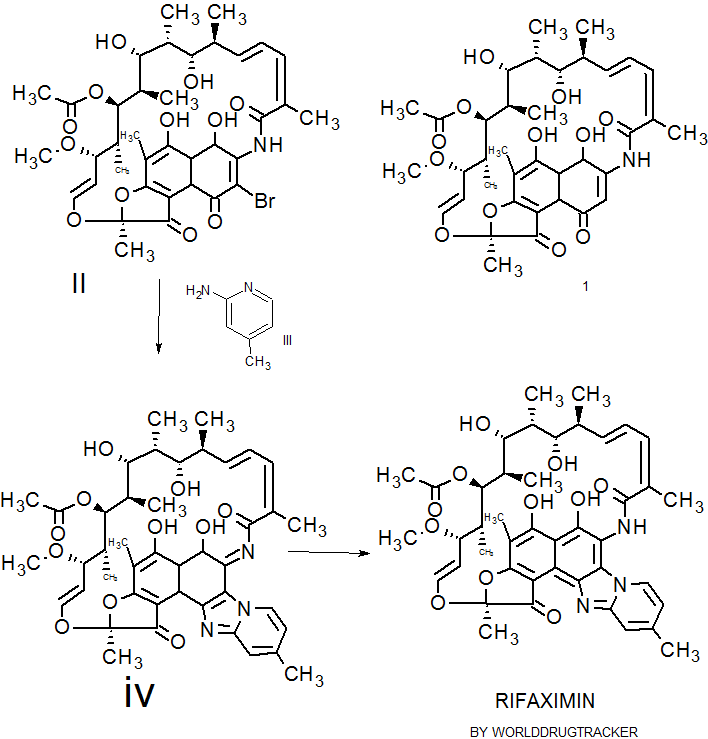
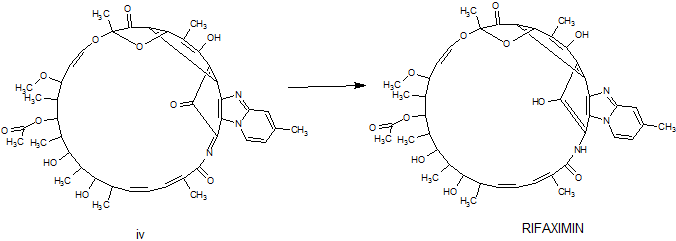
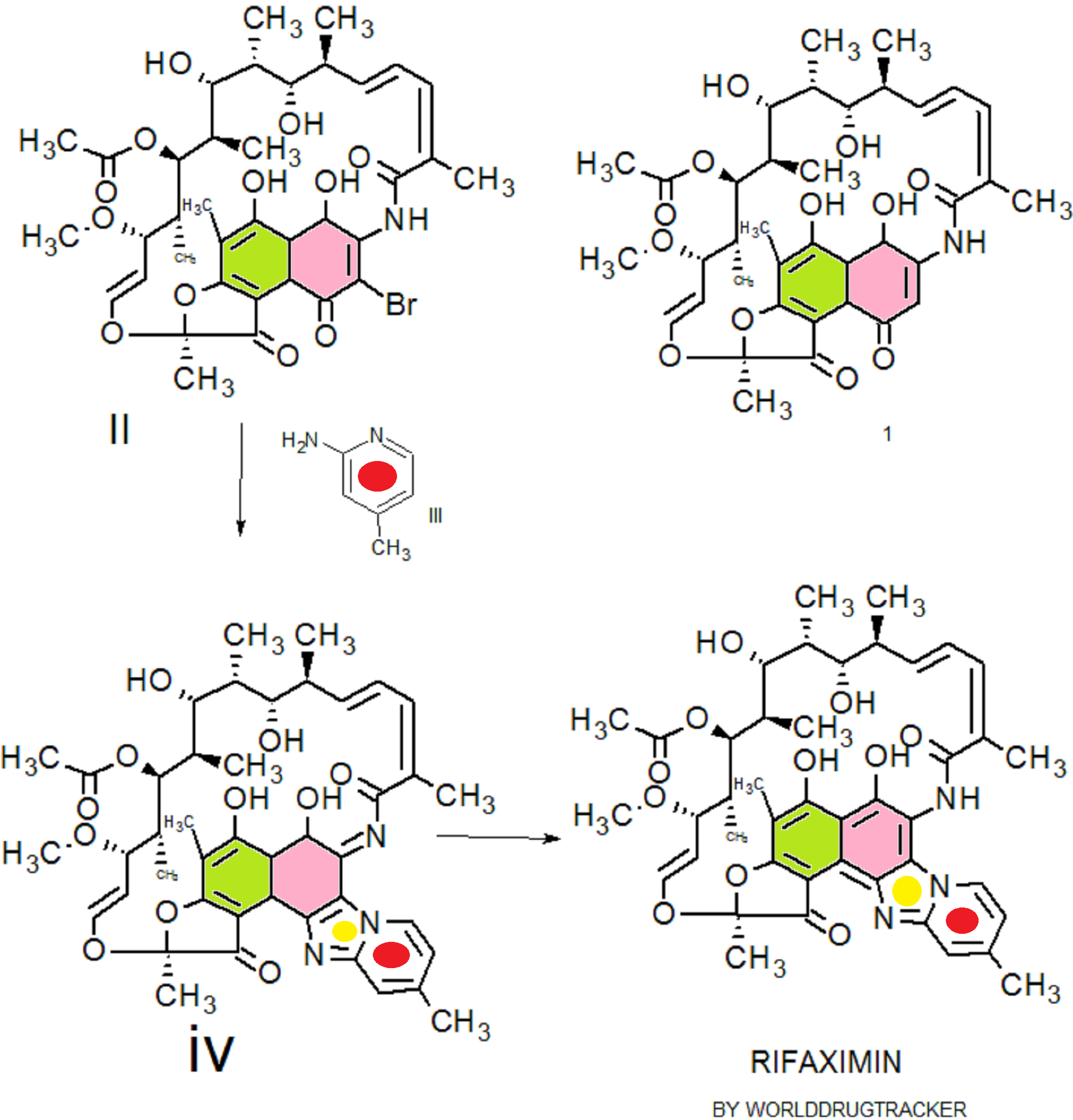
Rifaximin was first described and claimed in Italian patent IT 1154655 and U.S. Pat. No.4,341,785. These patents disclose a process for the preparation of Rifaximin and a method for the crystallisation thereof. The process for the preparation of Rifaximin is as depicted in scheme I given below:

Scheme -I
U.S. Pat. No. 4,179,438 discloses a process for the preparation of 3-bromorifamycin S which comprises reaction of rifamycin S with at least two equivalents of bromine, per one mole of rifamycin S in the presence of at least one mole of pyridine per each equivalent of bromine and in the presence of ethanol, methanol or mixtures thereof with water at a
temperature not above the room temperature. The process is shown in the scheme given below:

Rifamycin S 3-Bromo-Rifamycin-S
U.S. Patent No.4,557, 866 discloses a process for one step synthesis of Rifaximin from Rifamycin O, which is shown in scheme II given below:

Rifamycin O Rifaximin
Scheme -II
US '866 patent also discloses purification of Rifaximin by performing crystallization of crude Rifaximin from a 7:3 mixture of ethyl alcohol/water followed by drying both under atmospheric pressure and under vacuum. The crystalline form which is obtained has not been characterized.
U.S. Patent No. 7,045,620 describes three polymorphic forms α, β and γ of Rifaximin. Form a and β show pure crystalline characteristics while the γ form is poorly crystalline. These polymorphic forms are differentiated on the basis of water content and PXRD. This patent also discloses processes for preparation of these polymorphs which involve use of specific reaction conditions during crystallization like dissolving Rifaximin in ethyl alcohol at 45-65°C, precipitation by adding water to form a suspension, filtering suspension and washing the resulted solid with demineralized water, followed by drying at room temperature under vacuum for a period of time between 2 and 72 hours. Crystalline forms a and β are obtained by immediate filtration of suspension when temperature of reaction mixture is brought to 0°C and poorly crystalline form γ is obtained when the reaction mixture is stirred for 5-6 hours at 0°C and then filtered the suspension. In addition to above these forms are also characterized by specific water content. For a form water content should be lower than 4.5%, for β form it should be higher than 4.5% and to obtain γ form, water content should be below 2%.
U.S. Patent No. 7,709,634 describes an amorphous form of Rifaximin which is prepared by dissolving Rifaximin in solvents such as alkyl esters, alkanols and ketones and precipitating by addition of anti-solvents selected from hydrocarbons, ethers or mixtures thereof.
U.S. Patent No. 8,193,196 describes two polymorphic forms of Rifaximin, designated δ and ε respectively. Form δ has water content within the range from 2.5 to 6% by weight (preferably from 3 to 4.5%).
U.S. Patent No 8,067,429 describes a-dry, β-1, β-2, ε-dry and amorphous forms of Rifaximin.
U.S. Patent No. 8,227,482 describes polymorphs Form μ, Form π, Form Omicron, Form Zeta, Form Eta, Form Iota and Form Xi of Rifaximin.
International application publications WO 2008/035109, WO 2008/155728, WO 2012/035544, WO 2012/060675, and WO 2012/156533 describes various amorphous or poorly crystalline forms of Rifaximin.
These polymorphic forms are obtained under different experimental conditions and are characterized by XRPD pattern.
The polymorphic forms of Rifaximin obtained from the prior art methods have specific water content. Transition between different polymorphic forms of Rifaximin occurs by drying or wetting of the synthesized Rifaximin. Hence, it is evident from above that Rifaximin can exist in number of polymorphic forms, formation of these polymorphic forms depends upon specific reaction conditions applied during crystallization and drying.
Rifaximin is a semi-synthetic, rifamycin-based non-systematic antibiotic. It is chemically termed as (2S,16Z,18E,20S,21S,22R,23R,24R,25S,26 S,27S, 28E)-5,6,21,23,255-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro[4,5-e]pyrido[1,2-a]-benzimida-zole-1,15(2H)-dione,25-acetate (I).

Rifaximin is used for treatment of travelers' diarrhea caused by noninvasive strains of Escherichia coli.
Rifaximin was first disclosed in US4341785 which also discloses a process for its preparation and a method for crystallization of rifaximin using suitable solvents or mixture of solvents. However, this patent does not mention the polymorphism of rifaximin.
Canadian patent CA1215976 discloses a process for the synthesis of imidazo rifamycins which comprises reacting rifamycin S with 2-amino-4-methyl pyridine.
US4557866 discloses a process for preparation of rifaximin, but does not mention the polymorphs of rifaximin.
US7045620 discloses crystalline polymorphic forms of rifaximin which are termed as rifaximin α, rifaximin β and rifaximin γ. These polymorphic forms are characterized using X-ray powder diffraction. Further this patent mentions that γ form is poorly crystalline with a high content of amorphous component. This patent also discloses processes for preparation of these polymorphs which involve use of processes of crystallization and drying as disclosed in US4557866along with control of temperature at which the product is crystallized, drying process, water content thereof. Further, according to this patent, crystal formation depends upon the presence of water within the crystallization solvent.
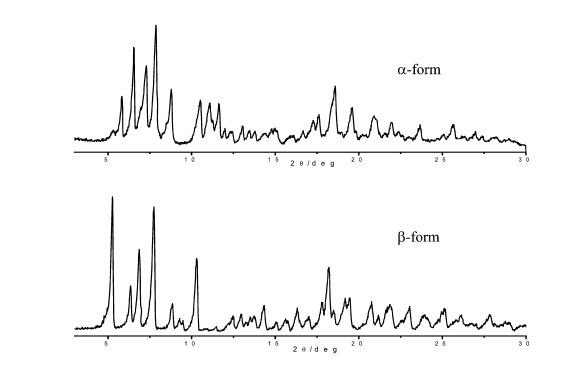
The above patent discloses rifaximin α which is characterized by water content lower than 4.5% & powder X-ray diffractogram having significant peaks are at values of diffraction angles 2θ of 6.6°; 7.4°; 7.9°, 8.8°, 10.5°, 11.1 °, 11.8°, 12.9°, 17.6°, 18.5°, 19.7°, 21.0°, 21.4°, 22.1°; rifaximin β which is characterized by water content higher than 4.5% & powder X-ray diffractogram having significant peaks are at values of diffraction angles 2θ of 5.4°; 6.4°; 7.0°, 7.8°, 9.0°, 10.4°, 13.1°, 14.4°, 17.1°, 17.9°, 18.3°, 20.9° and rifaximin γ which is characterized by poorer powder X-ray diffractogram because of poor crystallinity. The significant peaks are at values of diffraction angles 2θ of 5.0°; 7.1°; 8.4°.
US2005/0272754 also discloses polymorphs of rifaximin namely rifaximin α form, rifaximin β form & rifaximin γ form characterized by powder X-ray diffractogram, intrinsic dissolution rates and processes of preparation of polymorphic forms of rifaximin. However, none of the above patents disclose a wholly amorphous form of rifaximin.
It is a well known fact that different polymorphic forms of the same drug may have substantial differences in certain pharmaceutically important properties. The amorphous form of a drug may exhibit different dissolution characteristics and in some case different bioavailability patterns compared to crystalline forms.
Further, amorphous and crystalline forms of a drug may have different handling properties, dissolution rates, solubility, and stability.
Furthermore, different physical forms may have different particle size, hardness and glass transition temperatures. Amorphous materials do not exhibit the three-dimensional long-range orders found in crystalline materials, but are structurally more similar to liquids where the arrangement of molecules is random.
Amorphous solids do not give a definitive x-ray diffraction pattern (XRD). In addition, amorphous solids do not give rise to a specific melting point and tend to liquefy at some point beyond the glass transition temperature. Because amorphous solids do not have lattice energy, they usually dissolve in a solvent more rapidly and consequently may provide enhanced bioavailability characteristics such as a higher rate and extent of absorption of the compound from the gastrointestinal tract. Also, amorphous forms of a drug may offer significant advantages over crystalline forms of the same drug in the manufacturing process of solid dosage form such as compressibility.
| Drugs Fut 1982,7(4),260 | |
 | |
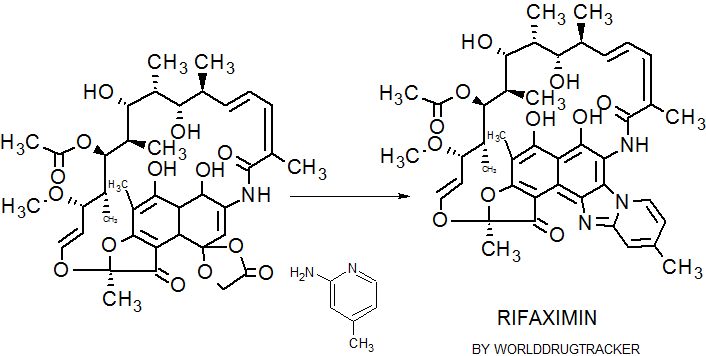
| The reaction of rifamycin S (I) with pyridine perbromide (II) in 2-propanol/chloroform (70/30) mixture at 0 C gives 3-bromorifamicin S (III), which is then condensed with 2-amino-4-methyl-pyridine (IV) at 10 C. The o-quinoniminic compound (V) is then obtained. This compound is finally reduced with ascorbic acid. | |
| US 262123 | |
| |
| The reaction of rifamycin S (I) with pyridine perbromide (II) in 2-propanol/chloroform (70/30) mixture at 0 C gives 3-bromorifamicin S (III), which is then condensed with 2-amino-4-methyl-pyridine (IV) at 10 C. The o-quinoniminic compound (V) is then obtained. This compound is finally reduced with ascorbic acid. | |
PATENT
The schematic representation for preparation of amorphous rifaximin is as follows :

Amorphous rifaximin according to the present invention can be characterized by various parameters like solubility, intrinsic dissolution, bulk density, tapped density.
Rifaximin is known to exist in 3 polymorphic Forms namely α Form, β Form & γ Form of which the α Form is thermodynamically the most stable. Hence, the amorphous form of rifaximin was studied in comparison with α Form.
Further, when intrinsic dissolution of amorphous rifaximin is carried out against the α Form, it is observed that the amorphous rifaximin has better dissolution profile than α Form which is shown in table below (this data is also shown graphically in Figure 3):
Dissolution medium : 1000 ml of 0.1M Sodium dihydrogen phosphate monohydrate + 4.5g of sodium lauryl sulphate
Temperature : 37±0.5°C
Rotation speed : 100 rpm
Particle size : Amorphous rifaximin - 11 microns
α Form of rifaximin - 13 microns
Time in minutes % Release of Amorphous Rifaximin % Release of α Form of Rifaximin 15 1.1 0.8 30 1.9 1.8 45 2.9 3.0 60 3.7 4.4 120 8.1 11.0 180 12.6 18.0 240 16.6 24.6 360 24.7 38.7 480 32.0 47.5 600 39.5 52.7 720 46.4 56.4 960 60.4 62.9 1200 72.9 67.8 1400 83.0 72.7 Amorphous rifaximin exhibits bulk density in the range of 0.3 - 0.4 g/ml and tapped density is in the range of 0.4 - 0.5 g/ml while the α Form rifaximin exhibits bulk density in the range of 0.2 - 0.3 g/ml & tapped density is in the range of 0.3 - 0.4 g/ml. These higher densities of amorphous rifaximin are advantageous in formulation specifically in tablet formulation, for example, it gives better compressibility.
CLIP
Rifaximin (CAS NO.: 80621-81-4), with other name of 4-Deoxy-4-methylpyrido[1,2-1,2]imidazo[5,4-c]rifamycin SV, could be produced through many synthetic methods.
Following is one of the reaction routes:

The reaction of rifamycin S (I) with pyridine perbromide (II) in 2-propanol/chloroform (70/30) mixture at 0 C gives 3-bromorifamicin S (III), which is then condensed with 2-amino-4-methyl-pyridine (IV) at 10 C. The o-quinoniminic compound (V) is then obtained. This compound is finally reduced with ascorbic acid.
POLYMORPHISM
Rifaximin (INN; see The Merck Index, XIII Ed., 8304) is an antibiotic belonging to the rifamycin class, exactly it is a pyrido-imidazo rifamycin described and claimed in Italian Patent IT 1154655, while European Patent EP 0161534 describes and claims a process for its production starting from rifamycin O (The Merck Index, XIII Ed., 8301).
Both these patents describe the purification of rifaximin in a generic way stating that crystallization can be carried out in suitable solvents or solvent systems and summarily showing in some examples that the reaction product can be crystallized from the 7:3 mixture of ethyl alcohol/water and can be dried both under atmospheric pressure and under vacuum without specifying in any way either the experimental conditions of crystallization and drying, or any distinctive crystallographic characteristic of the obtained product.
The presence of different polymorphs had just not been noticed and therefore the experimental conditions described in both patents had been developed with the goal to get a homogeneous product having a suitable purity from the chemical point of view, independent from the crystallographic aspects of the product itself.
It has now been found, unexpectedly, that there are several polymorphous forms whose formation, besides the solvent, depends on time and temperature conditions under which both crystallization and drying are carried out.
In the present application, these orderly polymorphous forms will be, later on, conventionally identified as rifaximin α (FIG. 1) and rifaximin β (FIG. 2) on the basis of their respective specific diffractograms, while the poorly crystalline form with a high content of amorphous component will be identified as rifaximin γ (FIG. 3).
Rifaximin polymorphous forms have been characterized through the technique of the powder X-ray diffraction.
The identification and characterization of these polymorphous forms and, simultaneously, the definition of the experimental conditions for obtaining them is very important for a compound endowed with pharmacological activity which, like rifaximin, is marketed as medicinal preparation, both for human and veterinary use. In fact it is known that the polymorphism of a compound that can be used as active ingredient contained in a medicinal preparation can influence the pharmaco-toxicologic properties of the drug. Different polymorphous forms of an active ingredient administered as drug under oral or topical form can modify many properties thereof like bioavailability, solubility, stability, colour, compressibility, flowability and workability with consequent modification of the profiles of toxicological safety, clinical effectiveness and productive efficiency.
What mentioned above is confirmed by the fact that the authorities that regulate the grant of marketing authorization of the drugs market require that the manufacturing methods of the active ingredients are standardized and controlled in such a way that they give homogeneous and sound results in terms of polymorphism of production batches (CPMP/QWP/96, 2003—Note for Guidance on Chemistry of new Active Substance; CPMP/ICH/367/96—Note for guidance specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances; Date for coming into operation: May 2000).
The need for the above-mentioned standardization has further been strengthened in the field of the rifamycin antibiotics by Henwood S. Q., de Villiers M. M., Liebenberg W. and Lotter A. P., Drug Development and Industrial Pharmacy, 26 (4), 403-408, (2000), who have ascertained that different production batches of the rifampicin (INN) made from different manufacturers differ from each other in that they show different polymorphous characteristics, and as a consequence they show different dissolution profiles, along with a consequent alteration of the respective pharmacological properties.
By applying the crystallization and drying processes generically disclosed in the previous patents IT 1154655 and EP 0161534 it has been found that under some experimental conditions a poorly crystalline form of rifaximin is obtained, while under other experimental conditions other polymorphic crystalline forms of Rifaximin are obtained. Moreover it has been found that some parameters, absolutely not disclosed in the above-mentioned patents, like for instance preservation conditions and the relative ambient humidity, have the surprising effect to determine the polymorph form.
The polymorphous forms of rifaximin object of the present patent application were never seen or hypothesized, while thinking that, whichever method was used within the range of the described condition, a sole homogeneous product would always have been obtained, irrespective of crystallizing, drying and preserving conditions. It has now been found that the formation of α, β and γ forms depends both on the presence of water within the crystallization solvent, on the temperature at which the product is crystallized and on the amount of water present in the product at the end of the drying phase. Form α, form β and form γ of rifaximin have then been synthesized and they are the object of the invention.
Moreover it has been found that the presence of water in rifaximin in the solid state is reversible, so that water absorption and/or release can take place in time in presence of suitable ambient conditions; consequently rifaximin is susceptible of transition from one form to another, also remaining in the solid state, without need to be again dissolved and crystallized. For instance polymorph α, getting water by hydration up to a content higher than 4.5%, turns into polymorph β, which in its turn, losing water by drying up to a content lower than 4.5%, turns into polymorph α.
These results have a remarkable importance as they determine the conditions of industrial manufacturing of some steps of working which could not be considered critical for the determination of the polymorphism of a product, like for instance the washing of a crystallized product, or the preservation conditions of the end product, or the characteristics of the container in which the product is preserved.
The above-mentioned α, β and γ forms can be advantageously used as pure and homogeneous products in the manufacture of medicinal preparations containing rifaximin.
As already said, the process for manufacturing rifaximin from rifamycin O disclosed and claimed in EP 0161534 is deficient from the point of view of the purification and identification of the product obtained; it shows some limits also from the synthetic point of view as regards, for instance, the very long reaction times, from 16 to 72 hours, not very suitable to an industrial use and moreover because it does not provide for the in situ reduction of rifaximin oxidized that may be formed within the reaction mixture.
Therefore, a further object of the present invention is an improved process for the industrial manufacturing of the α, β and γ forms of rifaximin, herein claimed as products and usable as defined and homogeneous active ingredients in the manufacture of the medicinal preparations containing such active ingredient.
PATENT
FIG. 1 is a powder X-ray diffractogram of rifaximin polymorphic form α.
FIG. 2 is a powder X-ray diffractogram of rifaximin polymorphic form β.
FIG. 3 is a powder X-ray diffractogram of rifaximin polymorphic form γ.

PATENT
Rifaximin (INN; see The Merck Index, XIII Ed., 8304, CAS no. 80621-81-4), IUPAC nomenclature (2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25 pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-(1,11,13)trienimino)benzofuro(4,5-e)pyrido(1,2,-a)benzimidazole-1,15(2H)-dione,25-acetate) is a semi-synthetic antibiotic belonging to the rifamycin class of antibiotics. More precisely rifaximin is a pyrido-imidazo rifamycin described in the Italian patent IT 1154655, whereas the European patent EP 0161534 discloses a process for rifaximin production using rifamycin O as starting material (The Merck Index, XIII Ed., 8301).
U.S. Pat. No. 7,045,620, US 2008/0262220, US 7,612,199, US 2009/0130201 and Cryst. Eng. Comm., 2008, 10 1074-1081 (2008) disclose new forms of rifaximin.
WO 2008/035109 A1 discloses a process to prepare amorphous rifaximin, which comprises reaction of rifamycin S with 2-amino-4 picoline in presence of organic solvent like dichloromethane, ethylacetate, dichloroethylene, chloroform, in an inert atmosphere. When water is added to the reaction mixture, a solid precipitate corresponding to amorphous rifaximin is obtained.
The process described in this document can be assimilated to a crash precipitation, wherein the use of an anti-solvent causes the precipitation of rifaximin without giving any information about the chemical physical and biological characteristics of the rifaximin obtained.
WO 2009/108730 A2 describes different polymorphous forms of rifaximin and also amorphous forms of rifaximin. Amorphous forms are prepared by milling and crash precipitation and with these two different methods the amorphous rifaximin obtained from these two different processes has the same properties.
FIG. 4: 13C-NMR spectrum of rifaximin obtained by spray drying process.

FIG. 5: FT-IR spectrum of rifaximin obtained by spray drying process.

Patent
WO 2015014984
Rifaximin, lUPAC name:
(2S,16Z,18E,20S,21 S,22H,23H,24H,25S,26S,27S,28£)-5,6,21 ,23,25-pentahydroxy- 27-methoxy-2,4,1 1 ,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1 ,1 1 ,13]-trienimmino)-benzofuro-[4,5-e]-pirido-[1 ,2-oc]-benzimidazol-1 , 15(2 -/)-dione,25-acetate, is the compound of formula (I):

Rifaximin is a broad-spectrum antibiotic belonging to the family of rifamycins, devoid of systemic activity. In view of its physicochemical properties, it is not adsorbed in the gastrointestinal tract and therefore exerts its antimicrobial action inside the gastrointestinal tract. Rifaximin therefore has applications in the treatment of diarrhoea and of microbial infections of the gastrointestinal tract typically caused by E. coli, a microorganism which, being incapable of passing through the mucosa of the gastrointestinal tract, remains in contact with the gastrointestinal fluids. Rifaximin also has applications for the treatment of irritable bowel syndrome, Crohn's disease, diverticulitis and for antibiotic prophylaxis preceding surgical operations on the intestines.
Rifaximin was obtained and described for the first time in the EP161534 starting from rifamycin O and 2-amino-4-picoline in the presence of ethanol/water and
ascorbic acid/HCI to obtain raw rifaximin which is then treated with Ethanol/water to obtain crystallized rifaximin.
Polymorphic forms of rifaximin, and processes for their synthesis and purification, are described in various documents of the known art.
Rifaximin K was firstly described in WO2012/156951 . Such a crystalline form resulted to be more stable in the presence of humidity than the other known crystalline forms of rifaximin, thus enabling the storage, even for prolonged periods. Such a polymorph was obtained by a process starting from rifaximin comprising the following steps: -suspending or dissolving rifaximin in a 1 ,2-dimethoxyethane based solvent, recovering the product and drying to remove said 1 ,2-dimethoxyethane based solvent. In one of the embodiments of the invention 1 ,2-dimethoxyethane is used as the unique solvent of rifaximin, in other 1 ,2-dimethoxyethane is described as used in combination of n-heptane, methanol, acetonitrile, R-COO-R1 esters wherein R and R1 are independently C3-C6 alkyl radicals, and C3-C7 alkyl ketones, ethanol, isopropanol and water.
J Antibiot 1984, 37(12): 1611
WO 2015014984
Rifaximin, lUPAC name:
(2S,16Z,18E,20S,21 S,22H,23H,24H,25S,26S,27S,28£)-5,6,21 ,23,25-pentahydroxy- 27-methoxy-2,4,1 1 ,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1 ,1 1 ,13]-trienimmino)-benzofuro-[4,5-e]-pirido-[1 ,2-oc]-benzimidazol-1 , 15(2 -/)-dione,25-acetate, is the compound of formula (I):
Rifaximin is a broad-spectrum antibiotic belonging to the family of rifamycins, devoid of systemic activity. In view of its physicochemical properties, it is not adsorbed in the gastrointestinal tract and therefore exerts its antimicrobial action inside the gastrointestinal tract. Rifaximin therefore has applications in the treatment of diarrhoea and of microbial infections of the gastrointestinal tract typically caused by E. coli, a microorganism which, being incapable of passing through the mucosa of the gastrointestinal tract, remains in contact with the gastrointestinal fluids. Rifaximin also has applications for the treatment of irritable bowel syndrome, Crohn's disease, diverticulitis and for antibiotic prophylaxis preceding surgical operations on the intestines.
Rifaximin was obtained and described for the first time in the EP161534 starting from rifamycin O and 2-amino-4-picoline in the presence of ethanol/water and
ascorbic acid/HCI to obtain raw rifaximin which is then treated with Ethanol/water to obtain crystallized rifaximin.
Polymorphic forms of rifaximin, and processes for their synthesis and purification, are described in various documents of the known art.
Rifaximin K was firstly described in WO2012/156951 . Such a crystalline form resulted to be more stable in the presence of humidity than the other known crystalline forms of rifaximin, thus enabling the storage, even for prolonged periods. Such a polymorph was obtained by a process starting from rifaximin comprising the following steps: -suspending or dissolving rifaximin in a 1 ,2-dimethoxyethane based solvent, recovering the product and drying to remove said 1 ,2-dimethoxyethane based solvent. In one of the embodiments of the invention 1 ,2-dimethoxyethane is used as the unique solvent of rifaximin, in other 1 ,2-dimethoxyethane is described as used in combination of n-heptane, methanol, acetonitrile, R-COO-R1 esters wherein R and R1 are independently C3-C6 alkyl radicals, and C3-C7 alkyl ketones, ethanol, isopropanol and water.
Paper
The synthesis of 4-deoxypyrido(1',2'-1,2)imidazo(5,4-c)rifamycin SV derivativesJ Antibiot 1984, 37(12): 1611
LAST STEP DEPICTED AGAIN
Treatment of rifamycin S (I) with Pyr·Br2 in 2-PrOH/CHCl3 gives 3-bromorifamycin S (II) (1), which upon cyclocondensation with 2-amino-4-methyl-pyridine (III) (1,2,3) in CHCl3 (2) or EtOH (3) yields imine derivative (IV). Finally, reduction of (IV) with L-(+)- ascorbic acid (1,2,3) in MeOH (2) or EtOH (3) provides the target rifaximin (1,2,3).
PATENT
WO 2005044823, WO 2012035544, WO 2015014984
Rifaximin is prepared by the cyclocondensation of rifamycin-O with 2-amino-4-picoline in a solvent mixture such as acetone, acetonitrile, EtOH, MIBK, propylene glycol, i-PrOH or t-BuOH and H2O at 50 °C or EtOH/aceone/H2O or optionally in the presence of I2 in CH2Cl2
PATENT
The process is shown in the scheme given below:

Rifamycin-S
3-halo-Rifamycin-S

Examples
Example 1;
5g of Rifamycin S, 3.1 gms of 2-amino-4-methyl pyridine, 0.45 g of iodine, 1.65 ml of acetic acid and 20ml of acetonitrile were charged in a clean and dry round bottom flask followed by stirring the resultant reaction mixture at about 30°C for about 30 hours. The reaction progress was monitored by TLC, after completion of reaction, the reaction mass was quenched by adding a mixture of 4.0g of ascorbic acid dissolved in 20 ml of water. The resultant reaction suspension was stirred at about 25°C for about 15mins. 25 ml of dichloromethane was charged and stirred for about 15mins. followed by separation of organic and aqueous phases. The aqueous phase was extracted with 25 ml of dichloromethane followed by separation of organic and aqueous phases. The organic phases were combined and distilled at below about 50°C to yield Rifaximin as residue. 11.25ml of purified water and 11.25ml of ethanol were charged to the residue and stirred at about 30°C for about 15 mins. The resultant reaction
suspension was heated to about 75°C and stirred for about 30mins. The resultant reaction solution further cooled to about 25 °C and stirred for about 2 hours followed by further cooling to about 5°C for about 3 hours. The solid precipitated was filtered and the solid was washed with a mixture of 2.5ml of ethanol and 2.5 ml of purified water. The solid obtained was dried at about 50°C for about 10 hours to afford 3 g. of Rifaximin as crystalline form. Purity by HPLC: 99.85 area %.
PAPER
European journal of medicinal chemistry (2015), 103, 551-62
Patent
Examples
Example 1 : Purification of Rifamycin S
Rifamycin S (500g) and Ethanol (1.5L) were stirred and refluxed for 1 hour. The reaction mixture was then cooled slowly to ambience, stirred at this temperature for 2 hour and filtered. The product dried in vacuum oven at 40 °C to obtain 475g of pure Rifamycin S showing the des acetyl impurity below to 0.6%.
Example 2: Preparation of rifaximin
Rifamycin S (300 g) was stirred in dichloromethane (900 ml) at room temperature for 15 minutes to get a clear solution and then 2-Amino-4-methyl pyridine (139.2g) was added at room temperature under nitrogen atmosphere. Iodine (57. Og) dissolved in dichloromethane (2100ml), was added drop wise in 30-45 minutes at room temperature. The reaction mass was stirred for 22-24 hours at 25-30 °C. After completion of the reaction, a 20% solution of L(-) ascorbic acid in water (300 ml) was added. The reaction mixture was stirred for 45-60 minutes at room temperature and then cooled to 10-15 °C. The pH of the resulting solution was adjusted to 1.5-2.0 with slow addition of dilute hydrochloric acid under stirring. The reaction mass was stirred for 15-20 minutes and layers were separated. The organic layer was washed with demineralized water (1500 ml), 10% sodium thiosulfate solution (1500 ml) and with demineralized water till pH was neutral. The solvent was distilled off under vacuum at 40-45 °C to get a residue which was taken in cyclohexane (1500 ml) and stirred for 1 hour. The resulting solid was filtered, washed with cyclohexane (300 ml) crystallized from a mixture of ethyl alcohol and water (600ml; 420ml ethyl alcohol and 180 ml water) to get 240g of crude rifaximin having purity 99.3% by HPLC.
Example 3: Preparation of rifaximin
Step-1: Preparation of crude rifaximin
Rifamycin S (300 g) was stirred in dichloromethane (900 ml) at room temperature for 15 minutes to get a clear solution and then 2-amino-4-methyl pyridine (139.2g) was added at room temperature under nitrogen atmosphere. Iodine (57. Og) dissolved in dichloromethane (2100ml), was added drop wise in 30-45 minutes at room temperature and was stirred for 22-24 hours. After completion of the reaction, a 20% solution of L (-) ascorbic acid in water (300 ml) was added and stirred for 45-60 minutes. The reaction mass was cooled to 10-15 °C and pH of the resulting solution was adjusted to 1.5-2.0 with slow addition of dilute hydrochloric acid under stirring. The reaction mass was stirred for 15-20 minutes and layers were separated and the organic layer was washed with demineralized water (1500 ml), with 10% sodium thiosulfate solution (1500 ml) and demineralized water till pH was neutral. The solvent was distilled off under vacuum at 40-45 °C to obtain a residue which was crystallized from a mixture of ethyl alcohol and water (378ml ethyl alcohol and 162 ml water) and dried at 35-40 °C to obtain 240g crude rifaximin having purity 98.8% by HPLC. Step-2: Purification of crude rifaximin
Crude rifaximin (240g) was stirred in dichloromethane (2400ml) at room temperature, a neutral alumina (240g) was added, stirred for 1 hour and filtered. The solvent was then distilled off and residue was treated with ethyl acetate (2400ml) and stirred to dissolution. The resulting residue was crystallized from a mixture of ethyl alcohol and water (302ml ethyl alcohol and 130ml water) and dried at 35-40 "C to obtain 192g of rifaximin having purity 99.8% by HPLC.
PATENT
PAPER

PATENTS
| US4341785 | May 11, 1981 | Jul 27, 1982 | Alfa Farmaceutici S.P.A. | Imidazo-rifamycin derivatives with antibacterial utility |
| US4557866 | Apr 26, 1985 | Dec 10, 1985 | Alfa Farmaceutici S.P.A. | Process for the synthesis of pyrido-imidazo rifamycins |
| US7045620 | Dec 5, 2003 | May 16, 2006 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in medicinal preparations |
| US7612199 | Jun 4, 2009 | Nov 3, 2009 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β, and γ of rifaximin |
| US7902206 | Mar 8, 2011 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β and γ of rifaximin | |
| US7906542 | May 13, 2008 | Mar 15, 2011 | Alfa Wassermann, S.P.A. | Pharmaceutical compositions comprising polymorphic forms α, β, and γ of rifaximin |
| US7915275 | Mar 29, 2011 | Alfa Wassermann, S.P.A. | Use of polymorphic forms of rifaximin for medical preparations | |
| US7923553 | Apr 12, 2011 | Alfa Wassermann, S.P.A. | Processes for the production of polymorphic forms of rifaximin | |
| US7928115 | Apr 19, 2011 | Salix Pharmaceuticals, Ltd. | Methods of treating travelers diarrhea and hepatic encephalopathy | |
| US8158644 | Apr 17, 2012 | Alfa Wassermann, S.P.A. | Pharmaceutical compositions comprising polymorphic forms α, β, and γ of rifaximin | |
| US8158781 | Mar 4, 2011 | Apr 17, 2012 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β and γ of rifaximin |
| US8193196 | Feb 27, 2006 | Jun 5, 2012 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20050272754 * | May 24, 2005 | Dec 8, 2005 | Alfa Wassermann S.P.A. | Polymorphic forms of rifaximin, processes for their production and uses thereof |
| Reference | ||
|---|---|---|
| 1 | Viscomi, G. C., et al., "Crystal forms of rifaximin and their effect on pharmaceutical properties", Cryst Eng Comm, 2008, 10, 1074-1081, (May 28, 2008), 1074-1081. | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9186355 | Mar 30, 2015 | Nov 17, 2015 | Novel Laboratories | Rifaximin crystalline forms and methods of preparation thereof |
| WO2008035109A1 * | Sep 24, 2007 | Mar 27, 2008 | Cipla Limited | Rifaximin |
| WO2009108730A2 * | Feb 25, 2009 | Sep 3, 2009 | Salix Pharmaceuticals, Ltd. | Forms of rifaximin and uses thereof |
| WO2011080691A1 * | Dec 27, 2010 | Jul 7, 2011 | Silvio Massimo Lavagna | Method for the production of amorphous rifaximin |
| EP1698630A1 * | Mar 3, 2005 | Sep 6, 2006 | ALFA WASSERMANN S.p.A. | New polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20080262220 * | May 13, 2008 | Oct 23, 2008 | Giuseppe Claudio Viscomi | Polymorphic forms alpha, beta and gamma of rifaximin |
| US20090082558 * | Sep 20, 2007 | Mar 26, 2009 | Apotex Pharmachem Inc. | Amorphous form of rifaximin and processes for its preparation |
REFERENCED BY
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015014984A1 * | Aug 1, 2014 | Feb 5, 2015 | Clarochem Ireland Ltd. | A process for preparing rifaximin k |
| CN103360357A * | Aug 7, 2013 | Oct 23, 2013 | 中国药科大学 | A simvastatin-gliclazide co-amorphous compound |
| US9359374 | Jun 13, 2013 | Jun 7, 2016 | Apotex Pharmachem Inc. | Polymorphic forms of rifaximin |
| US4341785 * | May 11, 1981 | Jul 27, 1982 | Alfa Farmaceutici S.P.A. | Imidazo-rifamycin derivatives with antibacterial utility |
| US4557866 * | Apr 26, 1985 | Dec 10, 1985 | Alfa Farmaceutici S.P.A. | Process for the synthesis of pyrido-imidazo rifamycins |
| US7045620 * | Dec 5, 2003 | May 16, 2006 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in medicinal preparations |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8518949 | Jun 4, 2012 | Aug 27, 2013 | Alfa Wassermann S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20140079783 * | Jul 3, 2013 | Mar 20, 2014 | Alfa Wassermann Spa | Pharmaceutical Compositions Comprising Rifaximin and Amino acids, Preparation Methods and Use Thereof |
| CN101836959A * | May 20, 2010 | Sep 22, 2010 | 山东达因海洋生物制药股份有限公司 | Method for preparing almost bitterless rifaximin dry suspension |
| CN103269587A * | Jun 3, 2011 | Aug 28, 2013 | 萨利克斯药品有限公司 | New forms of rifaximin and uses thereof |
| WO2011153444A1 * | Jun 3, 2011 | Dec 8, 2011 | Salix Pharmaceuticals, Ltd | New forms of rifaximin and uses thereof |
References
- Xifaxan label information PDF Retrieved November 15, 2008.
- DuPont, H (2007). "Therapy for and Prevention of Traveler's Diarrhea". Clinical Infectious Diseases 45 (45 (Suppl 1)): S78–S84. doi:10.1086/518155. PMID 17582576.
- Ruiz J, Mensa L, Pons MJ, Vila J, Gascon J (May 2008). "Development of Escherichia coli rifaximin-resistant mutants: frequency of selection and stability". Journal of antimicrobial chemotherapy 61 (5): 1016–9. doi:10.1093/jac/dkn078. PMID 18325895.
- Martinez-Sandoval F, Ericsson CD, Jiang ZD, Okhuysen PC, Romero JH, Hernandez N, Forbes WP, Shaw A, Bortey E, DuPont HL (Mar–Apr 2010). "Prevention of travelers' diarrhea with rifaximin in US travelers to Mexico.". J Travel Med. 17 (2): 111–7.doi:10.1111/j.1708-8305.2009.00385.x. PMID 20412178.
- Sharara A, Aoun E, Abdul-Baki H, Mounzer R, Sidani S, ElHajj I (2006). "A randomized double-blind placebo-controlled trial of rifaximin in patients with abdominal bloating and flatulence". Am J Gastroenterol 101 (2): 326–33. doi:10.1111/j.1572-0241.2006.00458.x.PMID 16454838.
- Antibiotic May Help Ease Irritable Bowel, Businessweek, January 05, 2011
- Small intestinal bacterial overgrowth in rosacea: clinical effectiveness of its eradication. Parodi A, Paolino S, Greco A, Drago F, Mansi C, Rebora A, Parodi A, Savarino V.
- Wolf, David C. (2007-01-09). "Hepatic Encephalopathy". eMedicine. WebMD. Retrieved 2007-02-15.
- Lawrence KR, Klee JA (2008). "Rifaximin for the treatment of hepatic encephalopathy".Pharmacotherapy 28 (8): 1019–32. doi:10.1592/phco.28.8.1019. PMID 18657018.Free full text with registration at Medscape.
- Kimer, Nina; Krag, Aleksander; Gluud, Lise L. (March 2014). "Safety, efficacy, and patient acceptability of Rifaximin for hepatic encephalopathy". Patient Preference and Adherence 8: 331–338. doi:10.2147/PPA.S41565. PMC 3964161. PMID 24672227. Retrieved 14 April 2016.
- http://formularyjournal.modernmedicine.com/formulary-journal/news/clinical/clinical-pharmacology/rifaximin-nonabsorbable-broad-spectrum-antibio?page=full
- http://www.drugbank.ca/drugs/DB01220
- Pimentel, Mark; Lembo, Anthony; Chey, William D.; Zakko, Salam; Ringel, Yehuda; Yu, Jing; Mareya, Shadreck M.; Shaw, Audrey L.; Bortey, Enoch (January 2011). "Rifaximin Therapy for Patients with Irritable Bowel Syndrome without Constipation". N Engl J Med364 (1): 22–32. doi:10.1056/NEJMoa1004409. PMID 21208106.
- Bass NM, Mullen KD, Sanyal A et al. (March 2010). "Rifaximin treatment in hepatic encephalopathy". N Engl J Med 362 (12): 1071–1081. doi:10.1056/NEJMoa0907893.PMID 20335583.
- Clark, Brian. "Rifaximin (Xifaxan) is a Promising Drug for the Treatment of Inflammatory Bowel Disease". Human Data Projct. Human Data Project. Retrieved 28 March 2016.
- http://www.salix.com/products/xifaxan550.aspx
- http://www.accessdata.fda.gov/scripts/cder/ob/docs/obdetail.cfm?Appl_No=022554&TABLE1=OB_Rx
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm448328.htm
- http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/GastrointestinalDrugsAdvisoryCommittee/UCM203248.pdf
- http://www.salix.com/news-media/news/previous-years-news/fda-approves-xifaxan%C2%AE-550-mg-tablets-for-reduction-in-risk-of-overt-hepatic-encephalopathy-he-recurrence.aspx
- http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2013_zaxine_161256-eng.php
External links
- FDA label approved for Xifaxan (PDF warning)
- Xifaxan200 (manufacturer's website)
| Patents |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
| (2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro [4,5-e]pyrido[1,2-a]-benzimida-zole-1,15(2H)-dione,25-acetate | |
| Clinical data | |
| Trade names | Xifaxan, Xifaxanta, Normix, Rifagut |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a604027 |
| Pregnancy category |
|
| Routes of administration | Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | < 0.4% |
| Metabolism | Hepatic |
| Biological half-life | 6 hours |
| Excretion | Fecal (97%) |
| Identifiers | |
| CAS Number | 80621-81-4 |
| ATC code | A07AA11 (WHO) D06AX11(WHO) QG51AA06 (WHO)QJ51XX01 (WHO) |
| PubChem | CID 6436173 |
| DrugBank | DB01220 |
| ChemSpider | 10482302 |
| UNII | L36O5T016N |
| KEGG | D02554 |
| ChEBI | CHEBI:75246 |
| ChEMBL | CHEMBL1617 |
| Chemical data | |
| Formula | C43H51N3O11 |
| Molar mass | 785.879 g/mol |
Giuseppe Viscomi, Manuela Campana, Dario Braga, Donatella Confortini, Vincenzo Cannata, Paolo Righi, Goffredo Rosini, “Polymorphic forms of rifaximin, processes for their production and uses thereof.” U.S. Patent US20050272754, issued December 08, 2005.
Title: Rifaximin
CAS Registry Number: 80621-81-4
CAS Name: (2S,16Z,18E,20S,21S,22R,23R,24R,25S,26R,27S,28E)-25-(Acetyloxy)-5,6,21,23-tetrahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca[1,11,13]trienimino)benzofuro[4,5-e]pyrido[1,2-a]benzimidazole-1,15(2H)-dione
Additional Names: 4-deoxy-4¢-methylpyrido[1¢,2¢-1,2]imidazo[5,4-c]rifamycin SV; rifamycin L 105; rifaxidin
Manufacturers' Codes: L-105
Trademarks: Fatroximin (Fatro); Flonorm (Schering-Plough); Normix (Alfa); Rifacol (Formenti); Xifaxan (Salix)
Molecular Formula: C43H51N3O11
Molecular Weight: 785.88
Percent Composition: C 65.72%, H 6.54%, N 5.35%, O 22.39%
Literature References: Nonabsorbable semisynthetic rifamycin antibiotic. Prepn: BE 888895; E. Marchi, L. Montecchi, US4341785 (1981, 1982 both to Alfa); E. Marchi et al., J. Med. Chem. 28, 960 (1985); and NMR study: M. Brufani et al., J. Antibiot.37, 1611 (1984). X-ray crystal structure: idem et al., ibid. 1623. In vitro and in vivo antibacterial activity: A. P. Venturini, E. Marchi,Chemioterapia 5, 257 (1986). Toxicological study: G. Borelli, D. Bertoli, ibid. 263. Clinical trial in travelers' diarrhea: R. Steffen et al., Am. J. Gastroenterol. 98, 1073 (2003). Review of activity, pharmacokinetics and clinical experience in gastrointestinal infections: J. C. Gillis, R. N. Brogden, Drugs 49, 467-484 (1995); D. B. Huang, H. L. DuPont, J. Infection 50, 97-106 (2005).
Properties: Red orange powder, mp 200-205° (dec). uv max: 232, 260, 292, 320, 370, 450 nm (E1%1cm 489, 339, 295, 216, 119, 159). Sol in alcohols, ethyl acetate, chloroform, toluene. Insol in water. LD50 orally in rats: >2000 mg/kg (Borelli, Bertoli).
Melting point: mp 200-205° (dec)
Absorption maximum: uv max: 232, 260, 292, 320, 370, 450 nm (E1%1cm 489, 339, 295, 216, 119, 159)
Toxicity data: LD50 orally in rats: >2000 mg/kg (Borelli, Bertoli)
Therap-Cat: Antibacterial.
Therap-Cat-Vet: Antibacterial.
Keywords: Antibacterial (Antibiotics); Ansamycins.
|
MORE.......
Rifaximin, alpha-0817185, L-105, Xifaxan, Lumenax, Flonorm, RedActiv, Rifacol, Normix
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
///////Rifaximin, Rifaxidin, Rifacol, Xifaxan, Normix, Rifamycin L 105, 80621-81-4, Rifaximin, alpha-0817185, L-105, Xifaxan, Lumenax, Flonorm, RedActiv, Rifacol, Normix
CC1C=CC=C(C(=O)NC2=C(C3=C(C4=C(C(=C3O)C)OC(C4=O)(OC=CC(C(C(C(C(C(C1O)C)O)C)OC(=O)C)C)OC)C)C5=C2N6C=CC(=CC6=N5)C)O)C