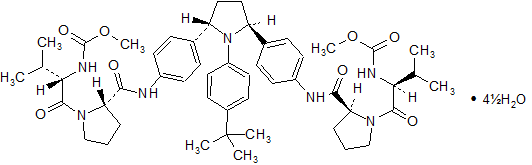
Ombitasvir Hydrate, 1456607-70-7

Ombitasvir 1258226-87-7
Ombitasvir; ABT-267; ABT 267; UNII-2302768XJ8; 1258226-87-7;
| C50H67N7O8 | |
| Molecular Weight: | 894.10908 g/mol |
|---|
Anti-Viral Compounds [US2010317568]
Methyl ((R)-1-((S)-2-((4-((2S,5S)-1-(4-(tert-butyl)phenyl)-5-(4-((R)-1-((methoxycarbonyl)-L-valyl)pyrrolidine-2-carboxamido)phenyl)pyrrolidin-2-yl)phenyl)carbamoyl)pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl)carbamate,
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate,
methyl N-[(2S)-1-[(2S)-2-[[4-[(2S,5S)-1-(4-tert-butylphenyl)-5-[4-[[(2S)-1-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]pyrrolidine-2-carbonyl]amino]phenyl]pyrrolidin-2-yl]phenyl]carbamoyl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate
| オムビタスビル水和物 Ombitasvir Hydrate  C50H67N7O8.4 1/2H2O : 975.18 [1456607-70-7] |
Abbvie Inc. innovator
Phase II clinical development at AbbVie (previously Abbott) for the treatment of chronic hepatitis C infection in combination with ABT-450/ritonavir and, in combination with peginterferon alpha-2a/ribavirin (pegIFN/RBV) in treatment naïve Hepatitis C virus (HCV) genotype 1 infected patients.
Phase II clinical development at AbbVie (previously Abbott) for the treatment of chronic hepatitis C infection in combination with ABT-450/ritonavir and, in combination with peginterferon alpha-2a/ribavirin (pegIFN/RBV) in treatment naïve Hepatitis C virus (HCV) genotype 1 infected patients.
Ombitasvir is Dimethyl ([(2S,5S)-1-(4-tert-butylphenyl) pyrrolidine-2,5diyl]bis{benzene-4,1-diylcarbamoyl(2S)pyrrolidine-2,1-diyl[(2S)-3-methyl-1-oxobutane-1,2diyl]})biscarbamate hydrate. The molecular formula is C50H67N7O8•4.5H2O (hydrate) and the molecular weight for the drug substance is 975.20 (hydrate).
Ombitasvir is in phase II clinical development at AbbVie (previously Abbott) for the treatment of chronic hepatitis C infection in combination with ABT-450/ritonavir and, in combination with peginterferon alpha-2a/ribavirin (pegIFN/RBV) in treatment naïve Hepatitis C virus (HCV) genotype 1 infected patients.
Ombitasvir is part of a fixed-dose formulation with ABT-450/ritonavir that is approved in the U.S. and the E.U.
In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. In 2013, breakthrough therapy designation was assigned in the U.S. for the treatment of genotype 1 hepatitis C in combination with ABT-450, ritonavir and ABT-333, with and without ribavirin.
Ombitasvir is in phase II clinical development at AbbVie (previously Abbott) for the treatment of chronic hepatitis C infection in combination with ABT-450/ritonavir and, in combination with peginterferon alpha-2a/ribavirin (pegIFN/RBV) in treatment naïve Hepatitis C virus (HCV) genotype 1 infected patients.
Ombitasvir is part of a fixed-dose formulation with ABT-450/ritonavir that is approved in the U.S. and the E.U.
In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. In 2013, breakthrough therapy designation was assigned in the U.S. for the treatment of genotype 1 hepatitis C in combination with ABT-450, ritonavir and ABT-333, with and without ribavirin.
Ombitasvir (Viekira PakTM) (Technivie )
Ombitasvir is an antiviral drug for the treatment of hepatitis C virus (HCV) infection. In the United States, it is approved by theFood and Drug Administration for use in combination with paritaprevir, ritonavir and dasabuvir in the product Viekira Pak for the treatment of HCV genotype 1,[1][2] and with paritaprevir and ritonavir in the product Technivie for the treatment of HCV genotype 4.[3][4]

Ombitasvir is an orally available inhibitor of the hepatitis C virus (HCV) non-structural protein 5A (NS5A) replication complex, with potential activity against HCV. Upon oral administration and after intracellular uptake, ombitasvir binds to and blocks the activity of the NS5A protein. This results in the disruption of the viral RNA replication complex, blockage of HCV RNA production, and inhibition of viral replication. NS5A, a zinc-binding and proline-rich hydrophilic phosphoprotein, plays a crucial role in HCV RNA replication. HCV is a small, enveloped, single-stranded RNA virus belonging to the Flaviviridae family; HCV infection is associated with the development of hepatocellular carcinoma (HCC).
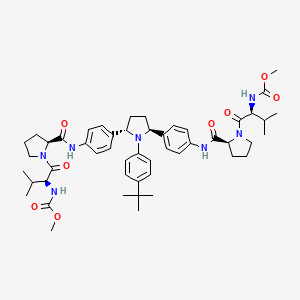

Ombitasvir hydrate is a NS5A non-nucleoside polymerase inhibitor which is approved as part of a four drug combination for the
treatment of adults with genotype 1 hepatitis C virus infection including those with compensated cirrhosis.REF 6,7
The four drug combination treatment consists of ombitasvir, paritaprevir (XXVII), ritonavir, and dasabuvir (X). This combination treatment is marketed as Viekira PakTM and was developed by Abbvie as an all oral treatment that eliminates the need for pegylated interferon-a injections.
The synthesis of ombitasvir hydrate is shown in Scheme 34.REF 8 Alkylation of 1-(4-nitrophenyl)ethanone (209)
with 2-bromo-1-(4-nitrophenyl)ethanone (208) in the presence of zinc chloride produced diketone 210 in 61% yield.
with 2-bromo-1-(4-nitrophenyl)ethanone (208) in the presence of zinc chloride produced diketone 210 in 61% yield.
Asymmetric reduction of the diketone using N,N-diethylaniline borane with (S)-( )-a,a-diphenyl-2-pyrrolidinemethanol (211) and trimethoxyborate gave diol 212 in 61% yield and 99.3% ee.
The diol was then treated with methanesulfonic anhydride to generate the corresponding bis-mesylate which was reacted with 4-tert-butylaniline to give pyrrolidine 213 in 51% yield over the two steps.
Hydrogenolysis of the nitro groups was accomplished using Raney nickel catalyst to give bis-aniline 214.
Separately, (L)-valine (216,Scheme 35) was reacted with methyl chloroformate to give the corresponding methyl carbamate in 90% yield which was coupled to L-proline benzyl ester in the presence of EDC and HOBt to give the corresponding dipeptide in 90% yield.
Hydrogenolysis of the benzyl ester group of the protected dipeptide using Pd/alumina catalyst produced dipeptide acid 215. Aniline 214 was treated with two equivalents of acid 215 in the presence of 1-propanephosphonic acid cyclic anhydride (T3P). The crude product was recrystallized from ethanol and heptane to give ombitasvir hydrate (XXV). No yields were provided to the final steps of this synthesis.

6 Gamal, N.; Andreone, P. Drugs Today (Barc) 2015, 51, 303.
7. DeGoey, D. A.; Randolph, J. T.; Liu, D.; Pratt, J.; Hutchins, C.; Donner, P.;Krueger, A. C.; Matulenko, M.; Patel, S.; Motter, C. E.; Nelson, L.; Keddy, R.;Tufano, M.; Caspi, D. D.; Krishnan, P.; Mistry, N.; Koev, G.; Reisch, T. J.;Mondal, R.; Pilot-Matias, T.; Gao, Y.; Beno, D. W.; Maring, C. J.; Molla, A.;Dumas, E.; Campbell, A.; Williams, L.; Collins, C.; Wagner, R.; Kati, W. M. J.
Med. Chem. 2014, 57, 2047.
8. DeGoey, D. A.; Kati, W. M.; Hutchins, C. W.; Donner, P. L.; Krueger, A. C.;Randolph, J. T.; Motter, C. E.; Nelson, L. T.; Patel, S. V.; Matulenko, M. A.;Keddy, R. G.; Jinkerson, T. K.; Soltwedel, T. N.; Liu, D.; Pratt, J. K.; Rockway, T.W.; Maring, C. J.; Hutchinson, D. K.; Flentge, C. A.; Wagner, R.; Tufano, M. D.;Betebenner, D. A.; Lavin, M. J.; Sarris, K.; Woller, K. R.; Wagaw, S. H.; Califano,
J. C.; Li, W.; Caspi, D. D.; Bellizzi, M. E. US Patent 2010317568A1, 2010.
Med. Chem. 2014, 57, 2047.
8. DeGoey, D. A.; Kati, W. M.; Hutchins, C. W.; Donner, P. L.; Krueger, A. C.;Randolph, J. T.; Motter, C. E.; Nelson, L. T.; Patel, S. V.; Matulenko, M. A.;Keddy, R. G.; Jinkerson, T. K.; Soltwedel, T. N.; Liu, D.; Pratt, J. K.; Rockway, T.W.; Maring, C. J.; Hutchinson, D. K.; Flentge, C. A.; Wagner, R.; Tufano, M. D.;Betebenner, D. A.; Lavin, M. J.; Sarris, K.; Woller, K. R.; Wagaw, S. H.; Califano,
J. C.; Li, W.; Caspi, D. D.; Bellizzi, M. E. US Patent 2010317568A1, 2010.
CLIP
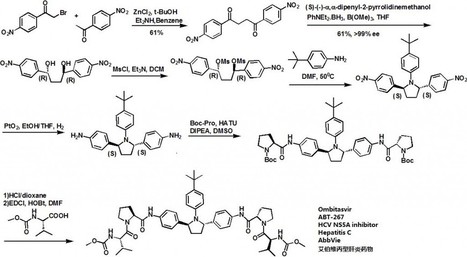
DeGoey, DA, Discovery of ABT-267, a Pan-genotypic Inhibitor of HCV NS5A, J. Med. Chem., 2014, 57 (5), pp 2047-2057
http://pubs.acs.org/doi/full/10.1021/jm401398x

We describe here N-phenylpyrrolidine-based inhibitors of HCV NS5A with excellent potency, metabolic stability, and pharmacokinetics. Compounds with 2S,5S stereochemistry at the pyrrolidine ring provided improved genotype 1 (GT1) potency compared to the 2R,5Ranalogues. Furthermore, the attachment of substituents at the 4-position of the central N-phenyl group resulted in compounds with improved potency. Substitution with tert-butyl, as in compound 38 (ABT-267), provided compounds with low-picomolar EC50 values and superior pharmacokinetics. It was discovered that compound 38 was a pan-genotypic HCV inhibitor, with an EC50 range of 1.7–19.3 pM against GT1a, -1b, -2a, -2b, -3a, -4a, and -5a and 366 pM against GT6a. Compound 38 decreased HCV RNA up to 3.10 log10 IU/mL during 3-day monotherapy in treatment-naive HCV GT1-infected subjects and is currently in phase 3 clinical trials in combination with an NS3 protease inhibitor with ritonavir (r) (ABT-450/r) and an NS5B non-nucleoside polymerase inhibitor (ABT-333), with and without ribavirin.
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate (38)...desired and Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2R,5R)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate (39).......undesired
................. The resulting mixture was stirred at room temperature for 16 h. The mixture was partitioned between ethyl acetate and water, and the organic layer was washed with saturated aqueous NaHCO3, brine (2×) and dried with Na2SO4. The drying agent was filtered off and the solution was concentrated in vacuo to give a crude product that was purified by column chromatography on silica gel, eluting with a solvent gradient of 2–8% methanol in dichloromethane to give a 1:1 mixture of trans-pyrrolidine isomers (290 mg, 96%). The mixture was separated on a Chiralpak AD-H column, eluting with a mixture of 1 part (2:1 isopropanol/ethanol) and 2 parts hexanes (0.1% TFA).
Compound 38 was the first of two stereoisomers to elute (101 mg, 99% ee by chiral HPLC). 1H NMR (400 MHz, DMSO-d6) δ 0.88 (d, J = 6.61 Hz, 6H), 0.93 (d, J = 6.72 Hz, 6H), 1.11 (s, 9H), 1.63 (d, J = 5.42 Hz, 2H), 1.80–2.04 (m, 8H), 2.09–2.19 (m, 2H), 2.44–2.47 (m, 2H), 3.52 (s, 6H), 3.59–3.66 (m, 2H), 3.77–3.84 (m, 2H), 4.02 (t, J = 8.40 Hz, 2H), 4.42 (dd, J = 7.86, 4.83 Hz, 2H), 5.14 (d, J = 6.18 Hz, 2H), 6.17 (d, J = 8.67 Hz, 2H), 6.94 (d, J = 8.78 Hz, 2H), 7.13 (d, J = 8.46 Hz, 4H), 7.31 (d, J= 8.35 Hz, 2H), 7.50 (d, J = 8.35 Hz, 4H), 9.98 (s, 2H).
MS (ESI) m/z 894.9 (M + H)+.
Compound39 was the second of two stereoisomers to elute. 1H NMR (400 MHz, DMSO-d6) δ 0.87 (d, J = 6.51 Hz, 6H), 0.92 (d, J = 6.72 Hz, 6H), 1.11 (s, 9H), 1.63 (d, J = 5.53 Hz, 2H), 1.82–2.04 (m, 8H), 2.09–2.18 (m, 2H), 2.41–2.47 (m, 2H), 3.52 (s, 6H), 3.58–3.67 (m, 2H), 3.75–3.84 (m, 2H), 4.02 (t, J = 7.26 Hz, 2H), 4.43 (dd, J = 7.92, 4.88 Hz, 2H), 5.14 (d, J = 6.18 Hz, 2H), 6.17 (d, J = 8.78 Hz, 2H), 6.94 (d, J = 8.67 Hz, 2H), 7.12 (d, J = 8.46 Hz, 4H), 7.31 (d, J = 8.35 Hz, 2H), 7.49 (d, J = 8.46 Hz, 4H), 9.98 (s, 2H). MS (ESI) m/z 895.0 (M + H)+.
PATENT
WO 2011156578
dimethyl (2S,2,S)-l,l '-((2S,2'S)-2,2'-(4,4'-((2S,5S)-l-(4-fert-butylphenyl)pyrrolidine- 2,5-diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3- methyl- l-oxobutane-2,l-diyl)dicarbamate

hereinafter Compound IA),..http://www.google.com/patents/WO2011156578A1?cl=en
PATENT
US 20100317568
Example 34
Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diyl)dicarbamate and
Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2R,5R)-1-(4-ter/'-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 - oxobutane-2, 1 -diyl)dicarbamate

Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4'-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2'S)-tert-Butyl 2,2'-(4,4'-((2S,5S)-1-(4-fer/'-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2'S)-tert-Butyl 2,2'- (4,4'-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig's base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2'S)-N,N'-(4,4'-((2S,5S)-1-(4-ter/'-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2'S)-N,N'-(4,4'-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diyl)dicarbamate and
Dimethyl (2S,2'S)-1, r-((2S,2'S)-2,2'-(4,4'-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 - oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 - 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 - 1.65 (m, 2 H) 1.79 - 2.04 (m, 8 H) 2.10 - 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 - 3.67 (m, 2 H) 3.76 - 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 - 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diyl)dicarbamate
 ..................desired
..................desired
The product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 - 2.04 (m, 8 H) 2.09 - 2.19 (m, 2 H) 2.44 - 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 - 3.66 (m, 2 H) 3.77 - 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2'S)-1, r-((2S,2'S)-2,2'-(4,4'-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 - oxobutane-2, 1 -diyl)dicarbamate
 .......undesired
.......undesired
The product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 - 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 - 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 - 3.67 (m, 2 H) 3.75 - 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diyl)dicarbamate
 ...............desired
...............desired
Example 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 - 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3x5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 - 2.04 (m, 8 H) 2.09 - 2.19 (m, 2 H) 2.44 - 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 - 3.66 (m, 2 H) 3.77 - 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 - 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
PATENT
Example 34
Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diyl)dicarbamate and
Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2R,5R)-1-(4-ter/'-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 - oxobutane-2, 1 -diyl)dicarbamate

Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4'-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2'S)-tert-Butyl 2,2'-(4,4'-((2S,5S)-1-(4-fer/'-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2'S)-tert-Butyl 2,2'- (4,4'-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig's base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2'S)-N,N'-(4,4'-((2S,5S)-1-(4-ter/'-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2'S)-N,N'-(4,4'-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diyl)dicarbamate and
Dimethyl (2S,2'S)-1, r-((2S,2'S)-2,2'-(4,4'-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 - oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 - 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 - 1.65 (m, 2 H) 1.79 - 2.04 (m, 8 H) 2.10 - 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 - 3.67 (m, 2 H) 3.76 - 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 - 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diyl)dicarbamate
 .............desired
.............desired
The product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 - 2.04 (m, 8 H) 2.09 - 2.19 (m, 2 H) 2.44 - 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 - 3.66 (m, 2 H) 3.77 - 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2'S)-1, r-((2S,2'S)-2,2'-(4,4'-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 - oxobutane-2, 1 -diyl)dicarbamate
 ..........undesired
..........undesired
The product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 - 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 - 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 - 3.67 (m, 2 H) 3.75 - 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diyl)dicarbamate
 ..................desired
..................desired
Example 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 - 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2'S)-l,r-((2S,2'S)-2,2'-(4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 - diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3x5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 - 2.04 (m, 8 H) 2.09 - 2.19 (m, 2 H) 2.44 - 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 - 3.66 (m, 2 H) 3.77 - 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 - 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Intermediates
Example 32
( 1 R,4R)- 1 ,4-bis(4-mtrophenyl)butane- 1 ,4-diol
To (S)-(-)-α,α-diphenyl-2-pyrrohdinemethanol (2 71 g, 10 70 mmol) was added THF (80 mL) at 23 °C The very thin suspension was treated with t11methyl borate (1 44 g, 13 86 mmol) over 30 seconds, and the resulting solution was mixed at 23 °C for 1 h The solution was cooled to 16-19 °C, and N,N-diethylanilme borane (21 45 g, 132 mmol) was added dropwise via syringe over 3-5 mm (caution vigorous H2 evolution), while the internal temperature was maintained at 16-19 °C After 15 mm, the H2 evolution had ceased To a separate vessel was added the product from Example IA (22 04 g, 95 wt%, 63 8 mmol), followed by THF (80 mL), to form an orange slurry After cooling the slurry to 11 °C, the borane solution was transferred via cannula into the dione slurry over 3-5 min During this period, the internal temperature of the slurry rose to 16 °C After the addition was complete, the reaction was maintained at 20-27 °C for an additional 2 5 h After reaction completion, the mixture was cooled to 5 °C and methanol (16 7 g, 521 mmol) was added dropwise over 5-10 mm, maintaining an internal temperature <20 °C (note vigorous H2 evolution) After the exotherm had ceased (ca 10 mm), the temperature was adjusted to 23 °C, and the reaction was mixed until complete dissolution of the solids had occurred Ethyl acetate (300 mL) and 1 M HCl (120 mL) were added, and the phases were partitioned The organic phase was then washed successively with 1 M HCl (2 x 120 mL), H2O (65 mL), and 10% aq NaCl (65 mL) The orgamcs were dried over MgSO4, filtered, and concentrated in vacuo Crystallization of the product occurred during the concentration The slurry was warmed to 50 °C, and heptane (250 inL) was added over 15 min. The slurry was then allowed to mix at 23 °C for 30 min and filtered. The wet cake was washed with 3: 1 heptane:ethyl acetate (75 mL), and the orange, crystalline solids were dried at 45 °C for 24 h to provide the title compound (15.35 g, 99.3% ee, 61% yield), which was contaminated with 11% of the meso isomer (vs. dl isomer).
References
- "VIEKIRA PAK™ (ombitasvir, paritaprevir and ritonavir tablets; dasabuvir tablets), for Oral Use. Full Prescribing Information"(PDF). AbbVie Inc., North Chicago, IL 60064. Retrieved 30 July 2015.
- "FDA approves Viekira Pak to treat hepatitis C". Food and Drug Administration. December 19, 2014.
- "TECHNIVIE™ (ombitasvir, paritaprevir and ritonavir) Tablets, for Oral Use. Full Prescribing Information" (PDF). AbbVie Inc., North Chicago, IL 60064. Retrieved 28 July 2015.
- "FDA approves Technivie for treatment of chronic hepatitis C genotype 4". Food and Drug Administration. July 24, 2015.
- Jordan J. Feld; Kris V. Kowdley; Eoin Coakley; Samuel Sigal; David R. Nelson; Darrell Crawford; Ola Weiland; Humberto Aguilar; Junyuan Xiong; Tami Pilot-Matias; Barbara DaSilva-Tillmann; Lois Larsen; Thomas Podsadecki & Barry Bernstein (2014). "Treatment of HCV with ABT-450/r–Ombitasvir and Dasabuvir with Ribavirin". N Engl J Med 370: 1594–1603. doi:10.1056/NEJMoa1315722.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015361087 | 2015-12-17 | ANTIVIRAL COMPOUNDS |
| US2015322108 | 2015-11-12 | CRYSTALLINE POLYMORPHS |
| US2015258093 | 2015-09-17 | SOLID ANTIVIRAL DOSAGE FORMS |
| US2015218194 | 2015-08-06 | Anti-Viral Compounds |
| US2015209403 | 2015-07-30 | Dose Adjustment |
| US2015196615 | 2015-07-16 | Methods for Treating HCV |
| US2015174194 | 2015-06-25 | METHODS FOR TREATING LIVER TRANSPLANT RECIPIENTS |
| US2015175646 | 2015-06-25 | SOLID FORMS OF AN ANTIVIRAL COMPOUND |
| US2015164976 | 2015-06-18 | Methods for Treating HCV |
| US2015150897 | 2015-06-04 | METHODS OF TREATING HEPATITIS C VIRUS INFECTION IN SUBJECTS WITH CIRRHOSIS |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015150897 | 2015-06-04 | METHODS OF TREATING HEPATITIS C VIRUS INFECTION IN SUBJECTS WITH CIRRHOSIS |
| US2015141351 | 2015-05-21 | Solid Pharmaceutical Compositions |
| US8993578 | 2015-03-31 | Methods for treating HCV |
| US8969357 | 2015-03-03 | Methods for treating HCV |
| US2015024999 | 2015-01-22 | Methods for Treating HCV |
| US2015011481 | 2015-01-08 | Methods for Treating HCV |
| US2014323395 | 2014-10-30 | Methods for Treating HCV |
| US2014315792 | 2014-10-23 | ANTI-VIRAL COMPOUNDS |
| US8853176 | 2014-10-07 | Methods for treating HCV |
| US8809265 | 2014-08-19 | Methods for treating HCV |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2014212491 | 2014-07-31 | COMBINATION FORMULATION OF TWO ANTIVIRAL COMPOUNDS |
| US2014171481 | 2014-06-19 | SOLID COMPOSITIONS |
| US8686026 | 2014-04-01 | Solid compositions |
| US8685984 | 2014-04-01 | Methods for treating HCV |
| US8680106 | 2014-03-25 | Methods for treating HCV |
| US2014080868 | 2014-03-20 | Methods for Treating HCV |
| US2014080869 | 2014-03-20 | Methods for Treating HCV |
| US2014080886 | 2014-03-20 | Methods for Treating HCV |
| US2014024613 | 2014-01-23 | Methods for Treating HCV |
| US8492386 | 2013-07-23 | Methods for treating HCV |
 | |
| Systematic (IUPAC) name | |
|---|---|
| Methyl ((R)-1-((S)-2-((4-((2S,5S)-1-(4-(tert-butyl)phenyl)-5-(4-((R)-1-((methoxycarbonyl)-L-valyl)pyrrolidine-2-carboxamido)phenyl)pyrrolidin-2-yl)phenyl)carbamoyl)pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl)carbamate | |
| Clinical data | |
| Trade names | Viekira Pak (with ombitasvir, paritaprevir, ritonavir and dasabuvir), Technivie (with ombitasvir, paritaprevir, and ritonavir) |
| Routes of administration | Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | not determined |
| Protein binding | ~99.9% |
| Metabolism | amide hydrolysis followed by oxidation |
| Onset of action | ~4 to 5 hours |
| Biological half-life | 21 to 25 hours |
| Excretion | mostly with feces (90.2%) |
| Identifiers | |
| CAS Number | 1258226-87-7 |
| PubChem | CID 54767916 |
| ChemSpider | 31136214 |
| ChEBI | CHEBI:85183 |
| Synonyms | ABT-267 |
| Chemical data | |
| Formula | C50H67N7O8 |
| Molar mass | 894.11 g/mol |
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 19 | |
|---|---|
| Patent | 8268349 |
| Expiration | Aug 25, 2024. 8268349*PED expiration date: Feb 25, 2025 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 2 of 19 | |
|---|---|
| Patent | 8466159 |
| Expiration | Sep 4, 2032 |
| Applicant | ABBVIE INC |
| Drug Application | N206619 (Prescription Drug: VIEKIRA PAK (COPACKAGED). Ingredients: DASABUVIR SODIUM ; OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 3 of 19 | |
|---|---|
| Patent | 9139536 |
| Expiration | Nov 9, 2028 |
| Applicant | ABBVIE INC |
| Drug Application | N206619 (Prescription Drug: VIEKIRA PAK (COPACKAGED). Ingredients: DASABUVIR SODIUM ; OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 4 of 19 | |
|---|---|
| Patent | 9044480 |
| Expiration | Apr 10, 2031 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 5 of 19 | |
|---|---|
| Patent | 9006387 |
| Expiration | Jun 10, 2030 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 6 of 19 | |
|---|---|
| Patent | 8691938 |
| Expiration | Apr 13, 2032 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 7 of 19 | |
|---|---|
| Patent | 8686026 |
| Expiration | Jun 9, 2031 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 8 of 19 | |
|---|---|
| Patent | 8685984 |
| Expiration | Sep 4, 2032 |
| Applicant | ABBVIE INC |
| Drug Application | N206619 (Prescription Drug: VIEKIRA PAK (COPACKAGED). Ingredients: DASABUVIR SODIUM ; OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 9 of 19 | |
|---|---|
| Patent | 8680106 |
| Expiration | Sep 4, 2032 |
| Applicant | ABBVIE INC |
| Drug Application | N206619 (Prescription Drug: VIEKIRA PAK (COPACKAGED). Ingredients: DASABUVIR SODIUM ; OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 10 of 19 | |
|---|---|
| Patent | 8642538 |
| Expiration | Sep 10, 2029 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 11 of 19 | |
|---|---|
| Patent | 8501238 |
| Expiration | Sep 17, 2028 |
| Applicant | ABBVIE INC |
| Drug Application | N206619 (Prescription Drug: VIEKIRA PAK (COPACKAGED). Ingredients: DASABUVIR SODIUM ; OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 12 of 19 | |
|---|---|
| Patent | 8492386 |
| Expiration | Sep 4, 2032 |
| Applicant | ABBVIE INC |
| Drug Application | N206619 (Prescription Drug: VIEKIRA PAK (COPACKAGED). Ingredients: DASABUVIR SODIUM ; OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 13 of 19 | |
|---|---|
| Patent | 8420596 |
| Expiration | Apr 10, 2031. 8420596*PED expiration date: Oct 10, 2031 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 14 of 19 | |
|---|---|
| Patent | 8399015 |
| Expiration | Aug 25, 2024. 8399015*PED expiration date: Feb 25, 2025 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 15 of 19 | |
|---|---|
| Patent | 8188104 |
| Expiration | May 17, 2029 |
| Applicant | ABBVIE INC |
| Drug Application | N206619 (Prescription Drug: VIEKIRA PAK (COPACKAGED). Ingredients: DASABUVIR SODIUM ; OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 16 of 19 | |
|---|---|
| Patent | 7364752 |
| Expiration | Nov 10, 2020. 7364752*PED expiration date: May 10, 2021 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 17 of 19 | |
|---|---|
| Patent | 7148359 |
| Expiration | Jul 19, 2019. 7148359*PED expiration date: Jan 19, 2020 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |
| FDA Orange Book Patents: 18 of 19 | |
|---|---|
| Patent | 6703403 |
| Expiration | Jun 26, 2016. 6703403*PED expiration date: Dec 26, 2016 |
| Applicant | ABBVIE INC |
| Drug Application | N207931 (Prescription Drug: TECHNIVIE. Ingredients: OMBITASVIR; PARITAPREVIR; RITONAVIR) |

/////Ombitasvir Hydrate, 1456607-70-7, Ombitasvir, 1258226-87-7, Viekira PakTM, Technivie , ABT-267, ABT 267, UNII-2302768XJ8, オムビタスビル 水和物 , phase II, clinical development , AbbVie, Abbott, chronic hepatitis C infection, combination with ABT-450/ritonavir, peginterferon alpha-2a/ribavirin (pegIFN/RBV), naïve Hepatitis C virus (HCV) genotype 1 infected patients.
O=C(Nc1ccc(cc1)[C@@H]5CC[C@@H](c3ccc(NC(=O)[C@@H]2CCCN2C(=O)[C@@H](NC(=O)OC)C(C)C)cc3)N5c4ccc(cc4)C(C)(C)C)[C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C
No comments:
Post a Comment