
Polmacoxib, CG-100649
(Acelex®)Approved
A COX-2 inhibitor used to treat osteoarthritis.
- OriginatorCrystalGenomics
- ClassAntirheumatics; Benzene derivatives; Fluorobenzenes; Furans; Nonsteroidal anti-inflammatories; Small molecules; Sulfonamides
- Mechanism of ActionCarbonic anhydrase inhibitors; Cyclo-oxygenase 2 inhibitors
- 12 Jan 2016Polmacoxib licensed to TR-Pharm for commercialisation in Turkey and Middle East and North Africa region
- 01 Sep 2015Launched for Osteoarthritis in South Korea (PO)
- 12 Aug 2015Polmacoxib licensed to Dong-A ST for commercialisation in South Korea
| Molecular Formula: | C18H16FNO4S |
|---|---|
| Molecular Weight: | 361.387343 g/mol |
CAS No.301692-76-2

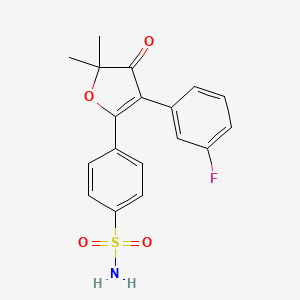
4-[3-(3-fluorophenyl)-5,5-dimethyl-4-oxofuran-2-yl]benzenesulfonamide

Polmacoxib (Acelex) is a nonsteroidal anti-inflammatory drug (NSAID) used to treat osteoarthritis. It was developed as CG100649 and approved for use in South Korea in February 2015.[1] It inhibits the enzymes carbonic anhydrase and COX-2. A study in healthy volunteers showed drug effects on urinary prostaglandin metabolites for both CG100649 and celecoxib that suggest a similar cardiovascular risk profile.[2] Further work by this group developed dose-exposure relationsships of CG100649 to guide clinical development strategies. [3]


| Dual-acting cyclooxygenase-2 (COX-2) and carbonic anhydrase inhibitor | |
| Molecular Target | Cyclooxygenase-2 (COX-2) ; Carbonic anhydrase l (CAI) |
| Mechanism of Action | Cyclooxygenase-2 (COX-2) inhibitor; NSAID |
KOREA FDA APPROVED ACELEX ® (POLMACOXIB) FOR THE TREATMENT OF OSTEOARTHRITIS
01 FEB
KOREA FDA APPROVED ACELEX ® (POLMACOXIB) FOR THE TREATMENT OF OSTEOARTHRITIS
CrystalGenomics, announced today that it has received approval for Acelex® (polmacoxib) from the Korean Ministry of Food and Drug Safety (MFDS) for the treatment of osteoarthritis.The company said that “Pre-commercialization will commence immediately and a commercial launch partner for the Korean market will be announced very shortly.”
Acelex® (polmacoxib) is the first, tissue-specific once-a-day osteoarthritis drug with a novel mode of action that specifically targets affected joints to relieve pain and restore mobility, while simultaneously preserving the integrity and safety of the gastrointestinal and cardiovascular systems. The results from the Phase 3 study suggest that Acelex 2mg once-a-day provides more rapid onset of relief from the signs and symptoms of osteoarthritis in comparison to celecoxib 200mg once-a-day, without added safety risk.
Polmacoxib is a first-in-class NSAID drug candidate that is a dual inhibitor of COX-2 and carbonic anhydrase (CA). Polmacoxib’s interaction with CA in red blood cells provides it with a novel ’tissue-specific’ transport mechanism that is designed to deliver sustained levels of drug to inflamed tissues, while maintaining low systemic exposure. Its unique dual COX-2 and CA binding properties are designed to provide potentially superior safety to cardiovascular, renal, and gastrointestinal tissues compared to traditional NSAIDs or COX-2 inhibitor drugs.
Acelex® is expected to rapidly capture at least 10% of the arthritis market in Korea that is estimated to be worth more than KRW 500 billion per year as of 2013. Osteoarthritis is quite common in Korea, as it affects about 50% of the population aged 65 years or older. Moreover, the overall number of patients is growing rapidly due to an aging population coupled with an increasing prevalence of obesity.
Nonsteroidal antiinflammatory drugs (NSAIDs) have been widely used over 100 years to alleviate symptoms of arthritis, arthritis-associated disorders, fever, post-operative pain, migraine, and so on. Despite their widespread use and desirable therapeutic efficacy for the treatment of inflammation and inflammation-associated disorders, NSAIDs are generally regarded to have life-threatening toxicity in the gastrointestinal (GI) tract. Severity of the GI toxicity is well illustrated by a report that 16 500 patients on NSAIDs therapy died due to the GI toxicity in the year of 1994 alone in the US. Frequently, the gastric toxicity of perforation, ulceration, and bleeding (PUB) is not noticed by patients before hospitalization, leading to such a high mortality rate upon chronic use of NSAIDs.
Despite the huge amount of efforts directed to reduce the GI toxicity of NSAIDs, it was only about a decade ago that the origin of the GI toxicity began to be understood through the discovery of an inducible isoform of cyclooxygenases. There are at least two kinds of cyclooxygenases, cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2). COX-1 is constitutively expressed in various tissues including the GI tract, the kidneys, and the platelets. COX-1 is known to be responsible for bodily homeostasis such as the gastrointestinal integrity, vascular dilatation, renal functions, and so on. Overt inhibition of COX-1 can, therefore, elicit undesirable side effects such as gastric PUB and blood thinning. In the meantime, COX-2 is induced upon inflammatory stimuli and is known to be responsible for progression of inflammation. Traditional NSAIDs, such as aspirin, naproxen, piroxicam, ibuprofen, diclofenac, etc., inhibit both COX-1 and COX-2, which accounts for NSAIDs' antiinflammatory effects as well as their notorious side effects of GI toxicity and blood thinning. Thus, selective inhibition of COX-2 over COX-1 should be useful for treatment of inflammation without incurring the side effects associated with inhibition of COX-1.
A study with COX-2 knockout mice suggests that complete inhibition of COX-2 could lead to peritonitis secondary to intestinal toxicity. Animal safety data for COX-2 inhibitors indicated that the intestinal toxicity was the dose-limiting toxicity in the dog and the rat. However, primates seem to possess robust intestinal tolerance to selective inhibition of COX-2. Indeed, COX-2 inhibitors are regarded to have better gastrointestinal safety profiles than traditional NSAIDs.
Long term use of traditional NSAIDs has been known to elicit cardiorenal toxicity such as edema and worsening blood pressure. There have been some attempts to assess cardiorenal safety of COX-2 inhibitors; however, more clinical data are needed to estimate the cardiorenal safety of COX-2 inhibitors. Considering that COX-2 inhibitors are supposed to be chronically taken mostlyby elderly arthritis patients, the importance of the long-term cardiorenal safety can never be overemphasized. COX-2 is constitutively expressed in the glomerular region and the small blood vessels of the kidneys in primates including the human, suggesting that the smaller inhibition of renal COX-2 could lead to smaller renal and consequently cardiovascular adverse effects. Given that only protein-unbound drug molecules are subject to glomerular filtration, a drug with higher plasma protein binding is expected to exert a smaller renal effect for a given lipophilicity or hydrophilicity of drug.
There are already several COX-2 inhibitors being prescribed for chronic indications, and they mostly maintain a tricyclic structure as in rofecoxib, celecoxib, valdecoxib, and etoricoxib.
Prostaglandins are known to play an important role in the inflammation.
Since prostaglandins are produced from arachidonic acid by cyclooxygenases, inhibition of prostagalndin synthesis by cyclooxygenases, especially synthesis of PGE2, PGG2, and PGH2, leads to the treatment of inflammation.
There are at least two kinds of cyclooxygenases, cyclooxygenase-1
(abbreviated as COX-1) and cyclooxygenase-2 (abbreviated as COX-2). COX-1 is constitutively present in the gastrointestinal tract and the kidney, and is implicated to be responsible for the maintenance of the physiological homeostasis, such as gastrointestinal integrity and renal function. Interruption of COX-1 activity can lead to life-threatening toxicities to the gastrointestinal tract, such as ulceration and bleeding. In the meantime, COX-2 is induced upon inflammatory stimuli and known to be responsible for progression of inflammation. Thus, selective inhibition of COX-2 over COX-1 is useful for the treatment of inflammation and inflammation-associated disorders without incurring gastrointestinal toxicities.
Conventional non-steroidal anti-inflammatory drugs (NSAIDs), such as indomethacin, naproxen, ketoprofen, ibuprofen, piroxicam, diclofenac etc, inhibit both COX-1 and COX-2, which would demonstrate their gastrointestinal toxicities as well as anti-inflammatory potency. However, they possess serious life-threatening gastrointestinal toxicities of bleeding and ulceration arising from their inhibition of COX-1, which limit their clinical use. Thus, a selective inhibitor of COX-2 can be useful as an anti-inflammatory therapeutic agent without the gastrointestinal toxicities, frequently occurring upon chronic use of conventional NSAIDs.
COX-2 inhibitors are implicated to possess a broad therapeutic spectrum besides anti-inflammatory, analgesic, and antipyretic activity. For example inhibition of COX-2 can prevent growth of certain types of cancer, especially colon cancer [J. Clin. Invest. 100. 1 (1997)]. Another application of a COX-2 inhibitor can be found in the treatment of degenerative chronic neurological disorders, such as Alzheimer's disease [Neurology 4£, 626 (1997)]. COX-2 inhibition would be useful in reducing the infarct volume accompanying the stroke [J. Neuroscience 17, 2746 (1997)].
Recently two of COX-2 selective antiinflammatory drugs, celecoxib and rofecoxib, were introduced into the clinic for arthritic indications. Celecoxib and rofecoxib show anti-inflammatory potency comparable to conventional NSAIDs without COX-2 selectivity. In the meantime, these drugs show appreciably lower gastrointestinal toxicities than conventional NSAIDs without COX-2 selectivity over COX-1. Thus, COX-2 selective inhibition itself can be enough for anti-arthritic potency and the inhibition of COX-1 is largely responsible for the gastro-intestinal toxicities associated with conventional NSAIDs without COX-2 selectivity.
.s-l,2-Diaryl-alkenes or its structural-equivalents are known to be a pharmacophore for achieving selective COX-2 inhibition over COX-1 [Ann. Rep. Med. Chem. 22, 211 (1997)]. In case of celecoxib and rofecoxib, pyrazole and 2(JH)-furanone correspond to the scaffold, respectively.

Celecoxib Rofecoxib By adopting an appropriate scaffold for the c/s-alkene pharmacophore, it would be possible to modulate both in vitro and in vivo characteristics of such inhibitors, such as dosing regimen, daily dose, clinical indications arising from tissue distribution characteristics, safety profile, and so on.
In this invention, 3(2H)-furanone is adopted as a scaffold for COX-2 inhibitors.
3(2H)-furanone derivatives were prepared for use in the treatment of glaucoma [EP 0
737 476 A2]. However, there is no precedent case that 3(2H)-furanone derivatives have been ever used as COX-2 inhibitors. There is no reported case of 4,5-diaryl-3(2H)-furanone derivatives, either.
The 4,5-diaryl-3(2H)-furanone derivatives disclosed herein selectively inhibit COX-2 over COX-1 and relieve the effects of inflammation. 4,5-Diaryl-3(2H)-furanone derivatives of this invention do not show substantial inhibition of COX-1 and consequently show reduced gastrointestinal side effects. Thus, 4,5-diaryl-3(2H)-furanone derivatives of this invention are found useful as anti -inflammatory agents with significantly reduced gastrointestinal side effects, when compared with conventional NSAIDs.
Since prostaglandins are produced from arachidonic acid by cyclooxygenases, inhibition of prostagalndin synthesis by cyclooxygenases, especially synthesis of PGE2, PGG2, and PGH2, leads to the treatment of inflammation.
There are at least two kinds of cyclooxygenases, cyclooxygenase-1
(abbreviated as COX-1) and cyclooxygenase-2 (abbreviated as COX-2). COX-1 is constitutively present in the gastrointestinal tract and the kidney, and is implicated to be responsible for the maintenance of the physiological homeostasis, such as gastrointestinal integrity and renal function. Interruption of COX-1 activity can lead to life-threatening toxicities to the gastrointestinal tract, such as ulceration and bleeding. In the meantime, COX-2 is induced upon inflammatory stimuli and known to be responsible for progression of inflammation. Thus, selective inhibition of COX-2 over COX-1 is useful for the treatment of inflammation and inflammation-associated disorders without incurring gastrointestinal toxicities.
Conventional non-steroidal anti-inflammatory drugs (NSAIDs), such as indomethacin, naproxen, ketoprofen, ibuprofen, piroxicam, diclofenac etc, inhibit both COX-1 and COX-2, which would demonstrate their gastrointestinal toxicities as well as anti-inflammatory potency. However, they possess serious life-threatening gastrointestinal toxicities of bleeding and ulceration arising from their inhibition of COX-1, which limit their clinical use. Thus, a selective inhibitor of COX-2 can be useful as an anti-inflammatory therapeutic agent without the gastrointestinal toxicities, frequently occurring upon chronic use of conventional NSAIDs.
COX-2 inhibitors are implicated to possess a broad therapeutic spectrum besides anti-inflammatory, analgesic, and antipyretic activity. For example inhibition of COX-2 can prevent growth of certain types of cancer, especially colon cancer [J. Clin. Invest. 100. 1 (1997)]. Another application of a COX-2 inhibitor can be found in the treatment of degenerative chronic neurological disorders, such as Alzheimer's disease [Neurology 4£, 626 (1997)]. COX-2 inhibition would be useful in reducing the infarct volume accompanying the stroke [J. Neuroscience 17, 2746 (1997)].
Recently two of COX-2 selective antiinflammatory drugs, celecoxib and rofecoxib, were introduced into the clinic for arthritic indications. Celecoxib and rofecoxib show anti-inflammatory potency comparable to conventional NSAIDs without COX-2 selectivity. In the meantime, these drugs show appreciably lower gastrointestinal toxicities than conventional NSAIDs without COX-2 selectivity over COX-1. Thus, COX-2 selective inhibition itself can be enough for anti-arthritic potency and the inhibition of COX-1 is largely responsible for the gastro-intestinal toxicities associated with conventional NSAIDs without COX-2 selectivity.
.s-l,2-Diaryl-alkenes or its structural-equivalents are known to be a pharmacophore for achieving selective COX-2 inhibition over COX-1 [Ann. Rep. Med. Chem. 22, 211 (1997)]. In case of celecoxib and rofecoxib, pyrazole and 2(JH)-furanone correspond to the scaffold, respectively.
Celecoxib Rofecoxib By adopting an appropriate scaffold for the c/s-alkene pharmacophore, it would be possible to modulate both in vitro and in vivo characteristics of such inhibitors, such as dosing regimen, daily dose, clinical indications arising from tissue distribution characteristics, safety profile, and so on.
In this invention, 3(2H)-furanone is adopted as a scaffold for COX-2 inhibitors.
3(2H)-furanone derivatives were prepared for use in the treatment of glaucoma [EP 0
737 476 A2]. However, there is no precedent case that 3(2H)-furanone derivatives have been ever used as COX-2 inhibitors. There is no reported case of 4,5-diaryl-3(2H)-furanone derivatives, either.
The 4,5-diaryl-3(2H)-furanone derivatives disclosed herein selectively inhibit COX-2 over COX-1 and relieve the effects of inflammation. 4,5-Diaryl-3(2H)-furanone derivatives of this invention do not show substantial inhibition of COX-1 and consequently show reduced gastrointestinal side effects. Thus, 4,5-diaryl-3(2H)-furanone derivatives of this invention are found useful as anti -inflammatory agents with significantly reduced gastrointestinal side effects, when compared with conventional NSAIDs.
Shin, Song Seok; Journal of Medicinal Chemistry 2004, V47(4), P792-804
In Vitro Structure−Activity Relationship and in Vivo Studies for a Novel Class of Cyclooxygenase-2 Inhibitors: 5-Aryl-2,2-dialkyl-4-phenyl-3(2H)furanone Derivatives
Drug Discovery, AmorePacific R&D Center, 314-1 Bora-ri, Kiheung-eup, Yongin-si, Kyounggi-do 449-729, South Korea
J. Med. Chem., 2004, 47 (4), pp 792–804
DOI: 10.1021/jm020545z

5-Aryl-2,2-dialkyl-4-phenyl-3(2H)furanone derivatives were studied as a novel class of selective cyclooxygenase-2 inhibitors with regard to synthesis, in vitro SAR, antiinflammatory activities, pharmacokinetic considerations, and gastric safety. 1f, a representative compound for methyl sulfone derivatives, showed a COX-2 IC50 comparable to that of rofecoxib. In case of 20b, a representative compound for sulfonamide derivatives, a potent antiinflammatory ED50 of 0.1 mg kg-1 day-1 was observed against adjuvant-induced arthritis by a preventive model, positioning20b as one of the most potent COX-2 inhibitors ever reported. Furthermore, 20b showed strong analgesic activity as indicated by its ED50 of 0.25 mg/kg against carrageenan-induced thermal hyperalgesia in the Sprague−Dawley rat. 3(2H)Furanone derivatives showed due gastric safety profiles as selective COX-2 inhibitors upon 7-day repeat dosing. A highly potent COX-2 inhibitor of the 3(2H)furanone scaffold could be considered suitable for a future generation COX-2 selective arthritis medication with improved safety profiles.

PATENT
non-steroidal anti-inflammatory drugs (nonsteroidal ant i- inf lammatory drug, NSAID) has a problem that causes serious side effects such as renal toxicity or distress Gastrointestinal. NSAID is to inhibit the activity of the enzyme cyclo-oxy-related prostaglandin G / H synthesis to tyrosinase (cyclooxygenase, COX) inhibits the biosynthesis of prostaglandins in the stomach and kidney, as well as inflammation. C0X is present in the two types of C0X C0X-1 and-2.
C0X-1 is induced by the other hand to adjust the height of the above features and is expressed in normal cells, it is C0X-2 mitogen or inflammation occurred in inflammation and other immune banung cytokines. To avoid the toxicity of the NSAID due to the inhibitory action of coexisting C0X-1 which, has been the selective inhibitors of the study C0X-2.
To 4- (3- (3-fluoro-phenyl) -5, 5-dimethyl-4-oxo-4, 5-dihydro-furan-2-yl) benzenesulfonamide represented by the general formula (1), such as 4, 5- diaryl-3- (0-furanones and derivatives thereof are compounds, wherein the by-1 do not inhibit the C0X standing substantially inhibit only C0X-2 selectively - represents a reduced gastrointestinal side effects while showing the inflammatory effect.

In addition, the compound of Formula 1 has C0X-2, as well as CA carbonic anhydrase) in inhibitory effect shown, in the CA-rich than C0X-2 tissues such as the gastrointestinal tract is to neutralize the inhibitory activity of C0X-2 gastrointestinal bleeding, such as side effects and more while the reduction, the less the distribution of the CA, such as joint tissue has a characteristic showing the effect to inhibit only C0X eu 2. Thus, 4, 5-diaryl-3- (0-furanones derivatives compared to conventional NSAIDs significantly reduced gastrointestinal side effects having an anti-inflammatory substance is useful as a.
Compounds and their derivatives of the formula (1) are of various inflammatory diseases; Pain accompanying diseases; viral infection; It is useful in the relief of inflammation, pain and fever, and the like accompanying surgery; diseases such as diabetes. Sikimyeo compounds and their derivatives of the formula (1) and they also inhibit the growth of cancer, including colorectal cancer C0X- parameter, reducing the infarction area of reperfusion injuries to (reperfusion injury) caused by the stroke, treatment of neurodegenerative diseases, including Alzheimer's disease it is useful. Diabetes accompanying retinopathy (retinopathy) in the treatment of useful and eu C0X-mediated vascularization (angiogenesis) to engage it (Mart in SG et al., Oral surgery oral medicine oral pathology, 92 (4), 2001, 399; James RH et al., oral surgery oral medicine oral pathology, 97 (2), 2004, 139; RE Harris et al., Inflammopharmacology, 12,2009, 55;
K. Oshima et al. , J. Invest. Surg. , 22 (4), 2009, 239; The Journal of
Pharmacology and Experimenral Therapeutics, 318 (3), 2006, 1248; JM. SL et al. , Int. J. Geriatr. Psychiatry, 2011; Jennifer L. et al. , Invest.
Ophthalmol. Vis. Sci. March, 44 (3), 2003, 974; K. M. Leahy et al. , Current Medicinal Chemistry, 7, 2000, 1163).
Method for producing a compound of formula I is disclosed in the International Patent Publication W0 00/615 sign, are incorporated herein by reference in their entirety.However, using the -78-butyllithium, which discloses in the above production method ° banung in C is not a m- chloroperoxybenzoic acid not suitable for commercial use it is difficult to practically carried out, as well as the yield for each step to be low, there are also overall yield is very low, so that problems 2.22%. "
Therefore, the way to mass production of a compound of formula 1 without problems, such as the high yield and a low cost has been desired still.
o provide the production method ol compound represented by Formula 1:



[Formula 4]
[Formula 5] 


[Formula 8] 
[Formula 9]

4- (3- (3-fluorophenyl) -5,5-dimethyl-4-oxo-4, 5-dihydro-furan-2-yl) -benzenesulfonamide The total yield by the method represented by Reaction Scheme 1 It is very easy to about 46% of the high yield and can be economically mass-produced:

Or less, on the basis of the example embodiments The invention will be described in more detail. The following examples are not be the only, and the scope of the invention to illustrate the present invention be limited to these.
Example 1: 2- (3-fluorophenyl) Preparation of the acetyl chloride

2- (3-fluorophenyl) acetic acid (305.5 g, 1.98 mol), thionyl chloride (500 mL, 6.85 mol) to dissolve by stirring the solution in a catalytic amount of dimethylformamide (2.1 mL, 25.83让ol) to the It was. This solution banung 110 ° and heated to sikimyeo C was stirred under reflux for 3 hours. After nyaenggak banung the solution to room temperature, the excess thionyl chloride under reduced pressure using a concentrator was removed by distillation. The stage was distilled off under reduced pressure to about 5mm¾ burgundy red oily objective compound (323 g, 94.4%) was obtained.
Example 2: 2- (3-fluorophenyl) -1- [4- (methylthio) phenyl] ethanone discussed prepared

Aluminum chloride (225 g, 1.91 mol) in dichloromethane (2500 mL), and then the suspension to 5 ° C a solution banung 2- (3-fluorophenyl) acetyl chloride (305 g in cooling,
It was added 1.77 mol). The reaction was stirred for about 5 minutes after the common compounds, the liquid Ndo of banung
5 ° while keeping the C was added dropwise the thio Enigma sol (237 g, 1.91 mol). After stirring for 3 hours banung common compounds at room temperature, it was slowly poured into cold aqueous hydrochloric acid solution. After separation the organic layer was washed with saturated aqueous sodium bicarbonate solution and brine and dried over anhydrous magnesium sulfate. After removing the anhydrous magnesium sulfate by filtration chest and diluted to a concentration under reduced pressure to concentrate the nucleic acid (1,000 mL). The diluted solution was 10 ° after the nyaenggak C to crystallize, it was stirred for 1 hour and then filtered and washed with a nucleic acid (1,000 mL). The filtered solid 50 ° and vacuum-dried for 2 hours in the target compound C (406 g, 88.3%) was obtained.
mp: 94.5 - 95.5 ° C
¾-NMR (CDCls, 300 MHz): δ 2.52 (s, 3H), 4.23 (s, 2H), 6.95-7.05 On, 3H), 7.25-7.30 (m, 3H), 7.92 (d, J = 8.7 Hz , 2H)
Example 3: 2,2-dimethyl-eu 4- (3-phenyl pool Luo) -5- [4- (methylthio) phenyl] -3 () - furanyl discussed prepared

Eu 2 (3-fluorophenyl) - 1- [4- (methylthio) phenyl] was cooled 30 minutes with stirring at ice-water was dissolved ethanone (512 g, 1.97 mol) in tetrahydrofuran (3,900 mL) . Sodium hydride in the reaction solution (60%, 180 g, 7.5 mol) was added to the subdivision for at least 15 minutes, the common banung compounds was stirred for 30 minutes at room temperature. The reaction common compounds 5 ° after nyaenggak in C, the 2-bromo butyryl cattle feeders cyanide (403 g, 2.29 mol) was added dropwise while maintaining the temperature. After the addition the solution was slowly stirred for 5 hours banung to room temperature. Banung ^ the compounds 5 ° and cooled to C, and then slowly added to de-ionized water and neutralized with acetic acid (122 g). After concentration under reduced pressure the banung solution was extracted with dichloromethane (2, 500 mL) and deionized water (2, 000 mL). The organic layer was washed with brine and then dried over anhydrous magnesium sulfate and filtered.
Filtered and concentrated under reduced pressure then gave a precipitate is dissolved with stirring in methanol (700 mL). After filtering the precipitate is washed with acid and methane. The filtered solid 50 ° and vacuum-dried for 2 hours at C, to give the desired compound (534.7 g, 82.8%). mp: 106 ° C
NMR-¾ (CDCI 3 , 300 MHz): δ 1.55 (s, 6H), 2.50 (s, 3H), 6.97-7.11 (m, 3H), 7.18 (d, J = 9.0 Hz, 2H), 7.26-7.36 (m, 1H), 7.55 ( d, J = 9.0 Hz, 2H)
Example 4: [4- (3- (3-fluoro-phenyl) -5, 5-dimethyl-4-oxo-4, 5-dihydro-furan-2-yl) phenylsulfonyl] Preparation of methyl acetate

2,2-dimethyl-eu eu eu 4 (3_ fluorophenyl) _5- [4- (methylthio) phenyl] -3 (0 furanones (5.5 Kg) and acetonitrile (27.2 Kg) and dichloromethane (45.43 Kg) after heunhap dissolved in a solvent, the compounds banung common -5 ° was cooled to C. to binary dissolved in acetic acid solution to the other reaction by injecting a peracetic acid (18%) and injection of dichloromethane and 23.4 Kg 13.9 Kg acetonitrile a common hapaek was prepared. hapaek prepared common to -5 ° keeping the C and slowly 0-5 was added to the reaction common compounds for 2 h ° and stirred for 30 to 90 minutes in the C. and the reaction common compounds with purified water 109.2 L separating the washed organic layer was then washed with aqueous sodium thiosulfate and aqueous sodium bicarbonate solution. the organic layer is concentrated 4- (3-fluorophenyl eu) eu 2,2-dimethyl-5- (4-eu
(Methyl sulfinyl) phenyl) furan -3 (2H) - one to give the as an oil form.
NiP: 143-144 ° C
¾-NMR (CDCls, 300 腿 ζ): δ 1.58 (s, 6Η), 2.76 (s, 3H), 7.26-7.08 (m, 3H), 7.30-7.38 (111, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 8.2 Hz, 2H)
After the thus obtained compound was dissolved in acetic anhydride (42.3 Kg) was added anhydrous sodium acetate (5.1 Kg). A liquid banung 130 ° under reflux for 12 hours at C and then cooled to room temperature after stirring. By filtration, washed with acetic anhydride solution banung the filtrate was 55 ° and concentrated in C. 63.5 Kg of purified water to the acid concentrate and 20.7
Injecting L and 10 ° after a nyaenggak C, it was added oxone 32.3 Kg followed by stirring for 3 hours. A liquid banung 50 ° and then concentrated in C until the residual liquid was added ½ and purified water (89.5 L) was stirred for 3 hours. The precipitated compound was filtered and then, washed with purified water and heptane and 50 °followed by drying for 12 hours at C, to give the desired compound (6.4 Kg, 91.3%).
¾ -赚(DMS0-d 6 (300 MHz): δ 8.01 (d, 2H), 7.83 (d, 2H), 7.43 (q, 1H), 7.20 (t, 1H), 7.07 (q, 1H), 5.47 (s, 2H), 2.06 ( s, 3H), 1.52 (s, 6H)
Example 5: Preparation of sodium 4- (3- (3-fluorophenyl) -5,5-dimethyl-4-oxo-4,5-dihydro-2-yl) Preparation of benzene sulfinate

[4- (3- (3-fluoro-phenyl) -5, 5-dimethyl-4-oxo-eu 4, 5-dihydro-furan-2-yl) phenylsulfonyl] methyl acetate (6.4 Kg) in tetrahydrofuran was dissolved in (34.3 Kg) and ethanol (15.3 Kg), the liquid temperature banung 0 ° was cooled to C. It was dissolved in sodium hydroxide (0.7 Kg) in purified water (16.1 L) to the other reaction section was prepared the solution cooled to C. It was added slowly for 5 hours, the prepared aqueous sodium hydroxide solution to the reaction solution, further stirring the reaction solution after about 1 hour and concentrated at 45 ° C. After concentration is completed, when added to absolute ethanol (10.0 Kg) and the toluene (11.0 Kg) was dissolved in concentrated 5C C. When concentration is complete, and then the absolute ethanol (10.0 Kg) was dissolved was added to toluene (10.1 Kg) and concentrated in 5C C. When the concentration is completed with absolute ethanol (7.7 Kg) was dissolved in 50 was added to toluene (8.4 Kg) ° was repeated in the course of concentration C twice. After re-concentrated solution of absolute ethanol (4.6 Kg) and the dissolution was added to toluene (5.1 Kg) to 50 ° and concentrated in C. Rouen (20.7 When the concentrate is completed,
Kg) was added and the resultant mixture was stirred for 2 hours, filtered and the washed with toluene (12.5 Kg). Was added to 20.7 Kg of toluene to the obtained solid was filtered after stirring for one to two hours. The filtered solid to a toluene (11.9 Kg) and washed with heptane (11.9 Kg) and then 45 ° was obtained in a quantitative and dried for 12 hours in C.
¾- 赚 (DMSO-de, 300 MHz): δ 7.52 (s, 4H), 7.40 (m, 1H), 7. 19-7.02
' (M, 3H), 1.49 (s, 6H) .
Example 6: 4- (3- (3-fluoro-phenyl) -5, 5-dimethyl-4-oxo-4, 5-dihydro-furan-2-yl) Preparation of benzenesulfonamide

Sodium 4- (3 eu (3_-fluorophenyl) -5, 5-dimethyl-4-oxo-4, 5-dihydro-furan-2-yl eudi) after the benzene sulfinate (6.0 Kg) was dissolved in dichloromethane - 5 ° and cooled to C. After stirring for another part banung ^ the combined dichloromethane (6.0 Kg) and sulfonic sulfuryl chloride (2. 1 Kg), 0 to the reaction solution obtained in the above ° was added slowly for 1 hour under C. A common banung hapaek eu 5 ° and after stirring for 4 hours at C and the organic layer was separated and washed with brine. After filtering the organic layer was dried over sodium sulfate (4.2 Kg), the filtrate was 40 ° and concentrated in C or less to give the intermediates of sulfonyl chloride compounds.
Tetrahydrofuran (36.3 Kg) and aqueous ammonia (16.9K the other part banung g were combined for common) was nyaenggak to 0 ° C. By dissolving the obtained sulfonic ponal chloride compound in 8.9 Kg of tetrahydrofuran 5 ° , while maintaining the below C was added slowly to the prepared aqueous ammonia solution for 1 hour.This solution banung -5 ° was concentrated after stirring for 30 to 120 minutes in the C. Once completed, the concentrated, purified water 40.2 L
It was added and stirred for 1 to 2 hours. Filtered and the resulting solid was then washed with purified water (16.9 L) and heptane (11.4 Kg). The filtered solid 45 °followed by drying for 12 hours at C, to give the desired compound (4.3 Kg, 73%).
mp: 204-205 ° C
¾-NMR (CDCls, 300 MHz): δ 1.57 (s, 6H), 4.96 (br s, 2H), 6.78 (m,
1H), 6.82 (m, 2H), 7.78 (d, J = 8.7 Hz, 2H), 7.96 (d, J = 8.7 Hz, 2H) IR (cm- 1 ): 3267, 1686, 1218, 1160
Example 7: Preparation of 2-bromo butyryl cattle feeders cyanide

Was added trimethylsilyl cyanide (283.4 g, 2.86 mol) in 2-bromo cattle feeders butyryl bromide (557 g, 2.24 mol). This solution banung 90 ° After stirring at C for 3 hours to nyaenggak to room temperature. Banung completed under reduced pressure (79画¾), 66 to 75 ° to fractional distillation under a C, to give the desired compound (384 g, 90.04%).
-醒(CDC1 3) 300 MHz): δ 1.97 (s, 6H)
PATENT

| Patent ID | Date | Patent Title |
|---|---|---|
| US2010069483 | 2010-03-18 | DUAL INHIBITION OF CYCLOOXYGENASE-2 AND CARBONIC ANHYDRASE |
| US2008306146 | 2008-12-11 | Dosing Regimens for Cox-2 Inhibitor |
| US2005222251 | 2005-10-06 | Dual inhibition of cyclooxygenase-2 and carbonic anhydrase |
| US6492416 | 2002-12-10 | 4,5-diaryl-3(2H)-furanone derivatives as cyclooxygenase-2 inhibitors |
| WO0061571 | 2000-10-19 | 4,5-DIARYL-3(2H)-FURANONE DERIVATIVES AS CYCLOOXYGENASE-2 INHIBITORS |
References
- "CrystalGenomics Receives MFDS Approval for Acelex® (Polmacoxib)". PR Newswire.
- Skarke, C.; Alamuddin, N.; Lawson, J. A.; Cen, L.; Propert, K. J.; Fitzgerald, G. A. (2012). "Comparative impact on prostanoid biosynthesis of celecoxib and the novel nonsteroidal anti-inflammatory drug CG100649". Clinical Pharmacology & Therapeutics 91 (6): 986–93. doi:10.1038/clpt.2012.3.PMC: 3740579. PMID 22278334.
- Hirankarn, S.; Barrett, J.S.; Alamuddin, N.; Fitzgerald, G. A.; Skarke, C. (2013). "GCG100649, A Novel Cyclooxygenase-2 Inhibitor, Exhibits a Drug Disposition Profile in Healthy Volunteers Compatible With High Affinity to Carbonic Anhydrase-I/II: Preliminary Dose–Exposure Relationships to Define Clinical Development Strategies". Clinical Pharmacology in Drug Development 2 (4): 379–386. doi:10.1002/cpdd.47.
 | |
| Systematic (IUPAC) name | |
|---|---|
| 4-(3-(3-Fluorophenyl)-5,5-dimethyl-4-oxo-4,5-dihydrofuran-2-yl)-benzenesulfonamide | |
| Clinical data | |
| Trade names | Acelex |
| Identifiers | |
| CAS Number | 301692-76-2 |
| PubChem | CID 9841854 |
| ChemSpider | 8017569 |
| UNII | IJ34D6YPAO |
| ChEMBL | CHEMBL166863 |
| Synonyms | CG100649 |
| Chemical data | |
| Formula | C12H16FNO4S |
| Molar mass | 361.3914 g/mol |
///////Polmacoxib, CG-100649, 301692-76-2
CC1(C(=O)C(=C(O1)C2=CC=C(C=C2)S(=O)(=O)N)C3=CC(=CC=C3)F)C
Plecanatide, also called Trulance, a synthetic analogue of uroguanylin, is a guanylate cyclase-C (GCC) agonist being developed by Synergy Pharmaceuticals for the treatment of gastrointestinal disorders, such as chronic idiopathic constipation (CIC) and irritable bowel syndrome with constipation (IBS-C). Plecanatide
ReplyDelete